Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Мышечная дистрофия: причины появления, симптомы, диагностика и способы лечения.
Определение
Мышечные дистрофии — это большая группа наследственных заболеваний, которые характеризуются прогрессирующей слабостью и дегенерацией скелетных мышц, то есть потерей мышечной массы. К наиболее распространенным миодистрофиям относятся миодистрофия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия), мышечная дистрофия Дюшенна и дистрофия Беккера. Также в эту группу входят дистрофия Эмери-Дрейфуса, миодистрофия Эрба-Рота и другие. Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей. Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них — Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
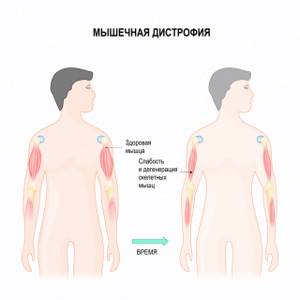 Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей. Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них — Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей. Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них — Х-сцепленной рецессивной формы, она составляет 1 на 100 000. Частота встречаемости миодистрофии Эрба-Рота составляет от 1,5 до 2,5 случаев на 100 тыс. населения. Этому типу мышечной дистрофии подвержены и мальчики, и девочки.
Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно, то есть патология проявляется, если ребенок получает аномальный ген от каждого из родителей. Каждый родитель может быть носителем дефектного гена, но обычно остается здоровым.
Около 30% генных мутаций возникают de novo, то есть не наследуются, а появляются «ниоткуда» и далее могут передаваться потомству.
Причины появления мышечной дистрофии
Причиной развития разных миодистрофий являются патологии в генах — известно порядка 25 генов, ответственных за развитие врожденных миодистрофий. При мышечной дистрофии Дюшенна вследствие мутации нарушается выработка белка дистрофина, который обеспечивает прочность, стабильность и функциональность мышечных волокон, и его нехватка приводит к повреждению мембран мышечных клеток (миоцитов).
Классификация заболеваний
Мышечные дистрофии могут классифицироваться в зависимости от того, какой белок подвергся мутации. Кроме того, их подразделяют по типу наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные.
Симптомы мышечной дистрофии
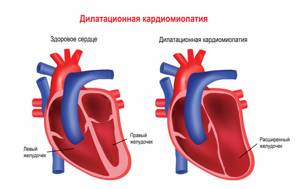
Мышечная дистрофия Дюшенна обычно манифестирует в возрасте 2-3 лет. Патологические процессы сначала происходят в мышцах ног, дети могут ходить на пальцах, вразвалку, отмечается избыточное выгибание позвоночника вперед – лордоз. Детям становится сложно бегать, прыгать, подниматься по лестнице, вставать с пола. Для разных типов миодистрофий характерным является симптом Говерса – вследствие слабости мышц бедер и тазового пояса больному, чтобы подняться из положения на корточках, приходится опираться руками об пол, затем подниматься, опираясь руками об колени. Мышечная слабость прогрессирует, у детей развивается сколиоз и сгибательные контрактуры – когда ребенок не может полностью разогнуть конечность. Дети часто падают, поэтому велик риск переломов рук или ног. Отдельные мышечные группы могут замещаться жировой или фиброзной тканью, в результате чего появляется псевдогипертрофия мышц, особенно заметная на лодыжках. Если страдает миокард (сердечная мышца), то существует предрасположенность к развитию нарушений ритма и проводимости сердца, а также дилатационной кардимиопатии (состояния, когда камеры сердца увеличены, а стенки истончены), приводящей к сердечной недостаточности.
К 12 годам большинство детей вынуждено пользоваться инвалидной коляской. В возрасте 15-20 лет пациентам уже требуется респираторная поддержка, умирают больные миодистрофией Дюшенна от дыхательных или кардиальных осложнений в возрасте 12-25 лет.
Симптомы миодистрофии Беккера обычно проявляются позже, и само заболевание протекает немного легче. Миодистрофия Беккера дебютирует в 10-20 лет и медленно прогрессирует, способность к самостоятельной ходьбе сохраняется в течение 15-20 лет от начала заболевания. Симптоматика схожа с миодистрофией Дюшенна, слабость распространяется на мышцы бедер, таза, плеч, пациенты ходят на носочках или вразвалку, также наблюдается гипертрофия мышц голеней. Развитие заболевания очень индивидуально, некоторым пациентам требуется инвалидное кресло к 30 годам, некоторые длительное время обходятся тростью. У пациентов с миодистрофией Беккера также отмечается поражение сердечной мышцы с развитием сердечной недостаточности. Первые признаки миодистрофии Ландузи-Дежерина проявляются в основном в возрасте 10-20 лет. Сначала атрофия и мышечная слабость наблюдаются в плечевом поясе с поражением мышц лопаток и плеч, потом распространяются на лицо с характерной асимметричностью. Начальными проявлениями являются затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании заболевания страдают лицевые мышцы, при этом пациент не может крепко зажмурить глаза и сжать губы. Позже мимика становится скудной, а речь неразборчивой. Характерными симптомами являются поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира»), «полированный» лоб. Иногда атрофия распространяется на мышцы ног. Другими клиническими признаками миодистрофии Ландузи-Дежерина могут быть аномалии сосудов сетчатки глаза, отек и отслойка сетчатки, снижение слуха. Для миодистрофии Эмери-Дрейфуса характерны контрактуры локтевых и голеностопных суставов, возникающие в раннем детстве (укорочение ахилловых сухожилий приводит к тому, что ребенок не может опуститься на пятки), тугоподвижность позвоночника, медленно прогрессирующая слабость лопаточно-плечевых и тазово-перонеальных мышц (мышц бедра и голени), а также выраженная кардиомиопатия с нарушениями ритма и проводимости. Тяжесть заболеваний сердца часто определяет прогноз течения болезни вследствие высокой вероятности внезапной сердечной смерти или развития прогрессирующей сердечной недостаточности. Миодистрофия Эрба-Рота сопровождается слабостью мышц поясничной области и конечностей. Первые признаки появляются в возрасте 10-20 лет: трудности при беге, быстрой ходьбе, прыжках, характерен симптом Говерса. Со временем начинает меняться осанка, походка, снижается тонус мышц плечевого пояса. С прогрессированием заболевания больной может полностью потерять способность ходить. Тотальная гипотрофия мышц туловища приводит к тому, что у пациента начинают выступать лопатки, талия становится очень тонкой, усиливается поясничный лордоз. Характерен симптом свободных надплечий — при попытке приподнять больного, удерживая его подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними.
Диагностика мышечной дистрофии
Предварительный диагноз врач может установить уже при осмотре, наблюдая за попытками ребенка побежать, прыгнуть, подняться по ступенькам, встать с пола. Для подтверждения диагноза проводятся:
- Анализ крови на сывороточную креатинкиназу, АСТ, АЛТ.
Креатинкиназа (Креатинфосфокиназа, КК, КФК, CK, Creatine kinaze)
Синонимы: Анализ крови на креатинкиназу. CK; Creatine Phosphokinase (CPK). Краткая характеристика определяемого вещества Креатинкиназа
Креатинкиназа катализирует обратимый перенос фосфорильного остатка с АТФ на креатин и с креатинфосфата на АДФ. Содержится преимущественно в скелетной мускулатуре…
АсАТ (АСТ, аспартатаминотрансфераза, AST, SGOT, Aspartate aminotransferase)
Синонимы: Глутамино-щавелевоуксусная трансаминаза; Глутамат-оксалоацетат-трансаминаза сыворотки крови (СГОТ); L-аспартат 2-оксоглутарат аминотрансфераза; ГЩТ. Aspartateaminotransferase; Serum Glutamicoxaloacetic Transaminase; SGOT; GOT. Краткая характеристика определяемого вещества АсАТ …
- мышечная дистрофия врожденная, тип 1C, FKRP м.;
Мышечная дистрофия врождённая, тип 1C, FKRP м.
Исследование мутаций в гене FKRP. Тип наследования. Аутосомно-рецессивный. Гены, ответственные за развитие заболевания. FKRP – ген фукутина (fukutin-related protein) Ген расположен на хромосоме 19 в локусе 19q13.3 и включает в себя четыре экзона, …
- мышечная дистрофия врожденная, тип 1C, FKRP ч.м.;
Мышечная дистрофия врождённая, тип 1C, FKRP ч.м.
Исследование частых мутаций в гене FKRP. Тип наследования. Аутосомно-рецессивный. Гены, ответственные за развитие заболевания. FKRP – ген фукутина (fukutin-related protein) Ген расположен на хромосоме 19 в локусе 19q13.3 и включает в себя четыре эк…
- прогрессирующая мышечная дистрофия Дюшенна/Беккера (ПМДД), DMD, делеции и дупликации экзонов 1-10, 21-30, 41-50, 61-70;
- мышечная дистрофия поясно-конечностная, FKRP м.;
Мышечная дистрофия поясно-конечностная, FKRP м.
Исследование мутаций в гене FKRP. Тип наследования. Аутосомно-рецессивный. Гены, ответственные за развитие заболевания. FKRP (FUKUTIN-RELATED PROTEIN) — фукутин-связанный белок. Расположен на хромосоме 19 в области 19q13.32. Содержит 3 некодирующих экзона…
- мышечная дистрофия тип Фукуяма, FKTN м.;
Мышечная дистрофия тип Фукуяма, FKTN м.
Исследование всех известных мутаций в гене FKTN. Тип наследования. Аутосомно-доминантный Гены, ответственные за развитие заболевания. FKTN (ген фукутина — FUKUTIN) Ген расположен на хромосоме 9 в области 9q31.2. Определение заболевания. …
- мышечная дистрофия Эмери-Дрейфуса, LMNA м.;
Мышечная дистрофия Эмери-Дрейфуса, LMNA м.
Исследование мутаций в гене LMNA. Тип наследования. Аутосомно-доминантный, аутосомно-рецессивный. Гены, ответственные за развитие заболевания. LMNA/C Ген ламина А/С расположен на хромосоме 1 в регионе 1q21.2-q21.3. включает 12 экзонов. В результате альте…
- мышечная дистрофия Эмери-Дрейфуса, FHL1 м.;
Мышечная дистрофия Эмери-Дрейфуса, FHL1 м.
Исследование мутаций в гене FHL1. Тип наследования. Х-сцепленный рецессивный. Гены, ответственные за развитие заболевания. Ген FHL1 (FOUR-AND-A-HALF LIM DOMAINS 1) расположен на Х-хромосоме в регионе Xq26.3. Содержит 5 экзонов. …
- мышечная дистрофия Эмери-Дрейфуса, ген эмерина при Х-сцепленной форме м.;
Мышечная дистрофия Эмери-Дрейфуса, ген эмерина при Х-сцепленной форме м.
Исследование мутаций в гене эмерина при Х-сцепленной форме. Тип наследования. Х-сцепленный рецессивный. Гены, ответственные за развитие заболевания. Ген эмерина (EMD) картирован на Х-хромосоме в области Xq28, состоит из 6 экзонов и 5 интро…
- мышечная дистрофия поясно-конечностная, SGCB м.;
Мышечная дистрофия поясно-конечностная, SGCB м.
Исследование мутаций в гене SGCB. Определение заболевания. Относится к группе прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового поясов конечностей. Относится к числу так называемых ламинопатий….
- мышечная дистрофия поясно-конечностная, SGCA м.;
Мышечная дистрофия поясно-конечностная, SGCA м.
Исследование мутаций в гене SGCA. Определение заболевания. Относится к группе прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового поясов конечностей. Относится к числу так называемых ламинопатий….
- мышечная дистрофия поясно-конечностная.
Мышечная дистрофия поясно-конечностная
Поиск частых мутаций в генах CAPN3, FKRP, ANO5, SGCA. Тип наследования. Аутосомно-рецессивный. Гены, ответственные за развитие заболевания. CAPN3 (calpain 3) — кальцием-активируемая нейтральная протеаза 3; ген находится на хромосоме …
- Для определения функции легких проводится исследование функции внешнего дыхания с определением форсированной жизненной емкости легких.
- Рекомендованы кардиологические обследования:
Эхокардиография
Исследование, позволяющее оценить функциональные и органические изменения сердца, его сократимость, а также состояние клапанного аппарата.
- До начала лечения стероидами проводят обследование на наличие антител к ветряной оспе и туберкулезу (в группах риска).
К каким врачам обращаться Заболевание может выявить педиатр, но для точной постановки диагноза потребуются консультации невролога и генетика.
- Лечение мышечной дистрофии
- Осложнения
- Профилактика мышечной дистрофии
- Источники:
Этиопатогенетическое лечение миодистрофий отсутствует. Поддерживающая терапия направлена на сохранение двигательной активности пациента и включает сбалансированную диету, обогащенную витаминами, микроэлементами, пищевые добавки, содержащие кальций, витамины группы В, Д, занятия ЛФК, массаж и физиопроцедуры. Для лечения мышечных дистрофий Дюшенна и Беккера применяют кортикостероидные гормоны, которые позволяют замедлить утрату мышечной силы и функций, уменьшить риск развития ортопедических осложнений и стабилизируют состояние легких и сердца. Вследствие поражения дыхательных мышц и миокарда у пациентов развиваются респираторные нарушения и сердечная недостаточность, которые и служат основной причиной смерти. Профилактикой мышечных дистрофий является медико-генетическое консультирование пар, планирующих зачатие ребенка, а также исключение близкородственных браков, в которых оба супруга могут быть носителями патологического гена. В качестве методов профилактики также рекомендована пренатальная диагностика плода у женщины-носительницы патологического гена.
- Narayanaswami P, Weiss M, Selcen D, David W, Raynor E, Carter G, Wicklund M, Barohn RJ, Ensrud E, Griggs RC, Gronseth G, Amato AA; Guideline Development Subcommittee of the American Academy of Neurology; Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2014 Oct 14;83(16):1453-63. doi: 10.1212/WNL.0000000000000892. PMID: 25313375; PMCID: PMC4206155.
Мышечная дистрофия Дюшенна
Этот тип мышечной дистрофии относится к прогрессирующим генетическим заболеваниям и характеризируется патогенными поражениями, некрозом мышечных волокон, замещением их жировой и соединительной тканью.
Симптоматика мышечной дистрофии Дюшенна
До 2-3 лет ребенок может не отставать в развитии от сверстников, первые симптомы проявляются на 3-5 году жизни, когда возникает мышечная слабость и утомляемость после физических нагрузок.
Отчетливое прогрессирование симптомов особенно заметно к 7-8 годам: меняется походка, а ослабленные мышцы начинают ограничивать самостоятельное передвижение больных. К 14-15 годам двигательная активность полностью ограничивается.
Характерными клиническими признаками мышечной дистрофии Дюшенна являются:
- симметричность развития патологических нарушений восходящего типа: вначале поражаются проксимальные мышц ног, таза и бедер, затем атрофия распространяется в область спины, плечевого пояса, верхних конечностей;
- при пальпации отмечается болезненность, плотность мышечных волокон;
- ограничиваются пассивные движения в суставах из-за непроизвольных сокращений мышечной ткани, возникают сухожильные ретракции;
- миодистрофия Дюшенна сочетается с патологическими изменениями костей и суставов (деформируются стопы, позвоночник), дисфункциональными расстройствами нейроэндокринной и сердечно-сосудистой систем;
- умственная отсталость проявляется в 30% случаев.
Этиология мышечной дистрофии Дюшенна
Возникновение и развитие патологии провоцирует генетическое поражение Х-хромосомы: нарушается синтез белка дистрофина, являющегося основой мышечных волокон и обеспечивающего их сокращение, пребывание в неактивном состоянии. При мышечной дистрофии этот белок не вырабатывается или продуцируется дефективным, что приводит к перерождению мышц и прогрессирующему ограничению двигательной активности.
Миодистрофия Дюшенна наследуется по рецессивному, сцепленному с Х-хромосомой типу. При этом девочки являются носителями патологии, а развивается заболевание только у лиц мужского пола (так как рецессивный аллель у них не подавляется доминантной парной генетически нормальной Х-хромосомой).
Диагностика мышечной дистрофии
- Аппаратные исследования: МРТ мышечной ткани применяется для определения степени поражений, ЭНМГ (электронейромиография) позволяет оценить функциональные характеристики периферической НС и мышц.
- Лабораторная диагностика: проводится биохимический АК на КФК (уровень креатинфосфокиназы) – увеличенная до 50 раз активность КФК при миодистрофических поражениях свидетельствует о прогрессировании заболевания.
Но наиболее показательными и информативными являются генетические молекулярные исследования (панель «Нервно-мышечные заболевания», микроматричный анализ – ХМА), которые точно определяют нозологическую форму мышечной дистрофии.
Кроме того, около трети диагностируемых случаев миодистрофии Дюшенна имеют спорадический характер. Возникновение 6-7% спонтанных мутаций обусловлено органным гонадным мозаицизмом – наличием у матери генетически здоровых и мутантных первичных половых клеток.
Поэтому пренатальная молекулярная диагностика позволяет идентифицировать, какой именно структурный вариант гена (аллель) получил ребенок. Даже если установлен мужской пол эмбриона, вероятность передачи от матери мутантной популяции гамет не оценивается в 100%, как и генетический риск наследования идентичной мутации братьями (сестрами) больного со спорадической миодистрофией.
Лечение мышечной дистрофии Дюшенна
- Медикаментозная терапия: прием ингибиторов АПФ улучшает состояние мышц, аталурен восстанавливает продуцирование дистрофина, гормональные препараты замедляют прогрессирование симптомов заболевания.
- Наиболее перспективной на сегодняшний день является генная терапия (экзон-скиппинг), которая дает возможность проводить коррекцию мутации, провоцирующей развитие мышечной дистрофии, с помощью добавления донорского экзогенного участка молекулы ДНК.
Мышечная дистрофия Дюшенна: причины, симптомы, лечение
Впервые псевдогипертрофическая прогрессирующая мышечная дистрофия была описана французским неврологом в середине девятнадцатого века — Гийомом Дюшенном, благодаря которому и получило свое название – дистрофия Дюшенна. Это одно из самых частых среди наследственных мышечно-дистрофических заболеваний, встречается с частотой 3-4 человека на сто тысяч населения.
Благодаря своей яркой клинической картине и достаточно широкой распространенности, многие люди впервые сталкиваясь с заболеванием (а также, к сожалению, часто и некоторые из врачей) считают, что есть только этот вид мышечной дистрофии, однако их множество и есть некоторые тонкости в симптоматике и диагностике заболевания. Об этом и пойдет речь в данной статье.
Информация для врачей. Все варианты наследственных мышечных дистрофий по МКБ10 шифруются в рубрике G71.0. Указывается тип заболевания по автору (может указываться также дифференциальный ряд, например, напоминающая дистрофию Дюшенна или Беккера). Также указывается стадия, скорость прогрессирования, степень слабости определенных групп мышц в баллах.
4 Лечение и прогноз жизни
Причины
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц.
Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна.
При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Симптомы
Начало развития симптомов начинается в раннем детском возрасте, чаще от 1 до 3 лет. Изначально отмечается отставание в моторном развитии, ребенок поздно начинает ходить, часто спотыкается при ходьбе, быстро устает. Позже развивается постоянная патологическая утомляемость мышц. Ребенок практически не может взбираться по лестнице. Походка начинает напоминать «утиную».
Постепенно начинает отмечаться атрофия мышц, вначале проксимальных отделов нижних, потом верхних конечностей. Позже атрофируются мышцы тазового пояса, бедер, спины, плечевого пояса. Почти всегда развивается «осиная» талия, искривление позвоночника, выпирание лопаток (крыловидные лопатки).
Можно выделить три стадии заболевания:
— I стадия – слабость проявляется лишь при значимой физической нагрузке (обычно первый год течения болезни).
— II стадия – затруднен подъем по лестнице, быстро развивается слабость при ходьбе.
— III стадия – представляет собой параличи, контрактуры мышц с невозможностью cсамостоятельного передвижения.
По видам течения подразделяется на:
Быстрое прогрессирование. Способность к передвижению утрачивается быстро, в течение первых 4-5 лет с начала заболевания.
Средний темп прогрессирования: передвигаться пациент не может спустя 10 лет.
Медленное прогрессирование: выраженных двигательных нарушений через 10 лет от начала болезни нет. Обычно такой вариант характерен для других типов мышечных дистрофий, нежели дистрофии Дюшенна.
Диагностика
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме. Обязательно необходимо проведение ЭМГ исследования для дифференциальной диагностики с другими нервно-мышечными заболеваниями.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. [!] В сложных диагностических ситуациях проводят цитологическое исследование.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики.
Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц.
Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.
Особенности диагностики прогрессирующей мышечной дистрофии дюшенна в детском возрасте — международный студенческий научный вестник (сетевое издание)
1
Шван Л.А. 1
Пацюра А.А. 1
Валиева А.Р. 1
Столярова Н.К. 1
Прогрессирующая мышечная дистрофия Дюшенна – наследственное заболевание, которое начинается в возрасте 2-5 лет и характеризуется прогрессирующей мышечной слабостью, атрофией и псевдогипертрофией проксимальных мышц, нередко сопровождается кардиомиопатиями и наруше-нием интеллекта.
При начале самостоятельной ходьбы отмечаются частые падения, спотыкания, мо-торная неловкость, повышенная утомляемость при ходьбе, происходит формирование «утиной по-ходки». Патогенез обусловлен мутацией в гене дистрофина, приводящей к прогрессирующей дегене-рации мышечных волокон.
При отсутствии дистрофина, мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Постепен-ная гибель мышечных волокон приводит к замещению их соединительнотканными структурами, ко-торые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии.
В данной статье тщательно изложены механизмы патогенеза и клинических симптомов при миодистрофии Дюшенна. Рассматриваются вопросы частоты распространения ПМДД. Так же авторы приводят клинический случай – пациент Х. мальчик 10 лет с подтвержденным результатом генетиче-ского исследования заболеванием ПМДД и характерными клиническими проявлениями.
Приводятся данные неврологического статуса и анамнеза пациента, так же основные клинические и лабораторно-инструментальные признаки при миодистрофии Дюшенна.
наследственные заболеванияпрогрессирующая мышечная дистрофия дюшенна
1. Николенко Н.Ю., Гончарова О.В., Артемьева С.Б., Ачкасов Е.Е., Литвнова Е.
Б Реабилитация детей с прогрессирующей мышечной дистрофией Дюшенна. Российский вестник перинатологии и педиатрии, 2014. №4. С.28-30 2. Деев Р.В., Мавликеев М.О., Бозо И.Я., Пулин А.А., Еремин И.И., Генноклеточная терапия наследственных заболеваний мышечной системы: современное состояние вопроса. //Гены & клетки. 2014. № 4. С.6-29
3. Абдрахманова Ж.
Клинико-диагностические аспекты верификации мышечной Дистрофии Дюшенна. //CLINICAL MEDICINE of KAZAKHSTAN. 2012. №4 (26) С. 97-99.
4. Mendell, J.R., and Lloyd-Puryear, M. Report of MDA muscle disease spoium on newborn screening for Duchenne muscular dystrophy. Annals of Neurology. 2012. vol. 71. no. 18. P. 304-313А.М.
5. Месова А.М.
Гипертрансфераземия при наследственных миопатиях у детей. // Вестник КазНМУ. 2016 – №2. С.115-117
Введение Прогрессирующая мышечная дистрофия Дюшенна (ПМДД) считается одним из распространенных наследственных заболеваний в неврологии. Болезнь наследуется по рецессивному, сцепленному с Х-хромосомой типу.
Дебют прогрессирующей мышечной дистрофии наступает в раннем возрасте, проявляется атрофией мышц, сочетающаяся с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением.
Существует два типа псевдогипертрофической мышечной дистрофии: тип Дюшенна с тяжелым течением, когда синтез дистрофина полностью блокирован, и доброкачественный тип Беккера, причиной которого является сниженная продукция дистрофина. Частота встречаемости ПМДД составляет 1 на 5000 новорожденных мальчиков.
В случае наличия кариотипа ХО или структурных аномалий хромосом (Хр21.2, ген DMD дистрофина), возможно, заболевание и у девочек, хотя встречается довольно редко.
Патоморфологическая особенность ПМДД – перерождение мышечной ткани, замещение ее жировой и соединительной тканью, а также некрозом отдельных волокон.
Начало заболевания характеризуется прогрессирующим нарастанием мышечной слабости сначала захватывает проксимальные мышцы нижних конечностей. Развивается характерная клиническая картина при которой у ребенка проявляется повышенная утомляемость при ходьбе, трудность при поднимании по лестнице, изменяется походка.
Распространяясь патологический процесс переходит на тазовый пояс, туловище, в меньшей степени, на плечевой пояс и проксимальные мышцы верхних конечностей. Отличительным симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотные, безболезненны.
Глубокие рефлексы изменяются с различной последовательностью, сначала исчезают коленные рефлексы, затем рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы длительное время остаются сохраненными. Атрофия мышц происходит всегда симметрично.
Как следствие атрофии появляются костно-суставные нарушения: поясничный гиперлордоз, кифоз, сколиоз, грудная клетка становится седловидной или килевидной появляется вальгус стопы. Формируются «осиная талия», крыловидные лопатки, симптом «свободных надплечий».
На рентгенограмме у пациента с ПМДД можно обнаружить сужение костномозгового канала, истончение коркового слоя длинных диафизов трубчатых костей. Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На электрокардиограмме (ЭКГ) регистрируются изменения миокарда (блокада ножек пучка Гиса и др.).
Течение ПМДД быстро прогрессирующее, злокачественное. К 7-10 годам возникают глубокие двигательные расстройства, выраженное изменение походки, снижение мышечной силы, что приводит к ограничению самостоятельного передвижения больных. А к 14-15 годам наступает обездвиженность. [1,2]
Доказано, что для ПМДД типично раннее, с 5-го дня жизни ребенка, повышение уровня мышечных энзимов, в частности повышение активности креатинфосфокиназы (КФК) — в 30-50 раз выше нормы, увеличение уровня печеночных трансаминаз, электромиографии и морфологии, выявляющих первично-мышечный тип поражения.
[3,4] Лабораторным критерием ПМДД является гипертрасфераземия, которая имеет внепеченочное происхождение. Повышенный выброс АлАТ и АсАТ обусловлен цитолизом мышечных клеток. По данным А.М.
Месовой (2016 год), среднее колебание значений КФК при ПМДД были в пределах от 5630 Е/л до 11000 Е/л, а АЛТ повышался от 256 МЕ/л до 551 МЕ/л, диапазон значений АсАТ составил от 128 МЕ/л до 286 МЕ/л.[5]
Целью нашего исследования является изучение клинико-лабораторных особенностей прогрессирующей мышечной дистрофии Дюшенна.
Материалы и методы: Нами описан клинический случай пациента Х., в возрасте 10 лет, с прогрессирующей мышечной дистрофии Дюшенна, находившегося на лечении в отделении КГП «ОДКБ». Данному ребенку были проведены комплексное обследование, консультация узких специалистов, консервативная, симптоматическая терапия и реабилитация.
Приводим клинический случай.
Пациент Х. 10 лет, 2008 года рождения. С диагнозом: Прогрессирующая мышечная дистрофия Дюшенна. Дискинезия желчевыводящих путей (ДЖВП) по гипотоническому типу. Реактивный панкреатит. Вторичная кардиомиопатия. МАРС (множественные дополнительные хорды левого желудочка). Недостаточность кровообращения (НД) 0-1 степени. Открытое овальное окно (ООО).
Anamnesis morbii: впервые ребенок заболел в возрасте 5 месяцев, когда впервые отмечались жалобы на вялость, слабость, неспособность удерживать голову из положения лежа на животе. При обращении в поликлинику по месту жительства с вышеуказанными жалобами была выявлена гипотония всех групп мышц.
Получал стационарное лечение в возрасте 9 месяцев по поводу перинатальной энцефалопатии, митотического синдрома. В амбулаторной карте описана задержка моторного развития: общая мышечная гипотония, вялая опора, шаговые движения не вызываются. В 1 год 5 месяцев отмечалась шаткость походки, частые падения, неловкость. В неврологическом статусе описано снижение сухожильных рефлексов.
В возрасте 1 год 7 месяцев ребенок не мог самостоятельно садиться, подняться из положения сидя, стал подниматься «лесенкой», при ходьбе переваливаться, отмечалась псевдогипертрофия обеих икроножных мышц. С 2009 по 2011 год за пациентом осуществлялось динамическое наблюдение у невропатолога по месту жительства с диагнозом миотонический синдром.
В 2012 году в связи со сменой лечащего врача были направлены на молекулярно-генетическое исследование в Центр молекулярной медицины г. Алматы.
В 2012 году, в возрасте 4-х лет, проведено молекулярно-генетическое исследование ДНК в Центре молекулярной медицины г. Алматы, на делеции во фрагментах 19 различных экзонов промоторной области, гена дистрофина (МПА1, МПА2), вызывающих миодистрофию Дюшенна/Беккера.
Миопатия Дюшенна
Главная » Болезни » Лечение опорно-двигательного аппарата » Миопатия
Миопатия – это хроническая прогрессирующая патология врожденного (наследственного) или приобретенного происхождения, обусловленная нарушением обмена веществ в мышечной ткани. Данное дегенеративное расстройство, характеризующееся прогрессирующей атрофией мышц, является собирательным термином, включающим в себя различные формы спинальных и невральных поражений скелетной мускулатуры.
Причины возникновения и развития миопатий
Основная причина миопатий – нарушение биохимических процессов в поперечно-полосатой мышечной ткани, приводящее к повышению уровня креатинфосфокиназы (фермента, обеспечивающего энергией мускульное сокращение).
Невозможность удержания и связывания энергетического посредника, креатинина, приводит к уменьшению содержания АТФ, распадающейся с выделением энергии, необходимой для обеспечения двигательной активности и сократительной способности.
Подобное состояние влечет за собой атрофию и в конечном итоге повреждение и гибель мышечных волокон.
Формы миопатии
Все известные медицине миопатии условно подразделяются на 2 обширные группы: врожденные и приобретенные.
Наследственные мышечные дистрофии:
- миопатия Дюшенна (самая распространенная форма);
- миопатия Беккера (доброкачественная);
- конечно-поясная миодистрофия Эрба −Рота;
- болезнь Верднига − Гоффмана;
- дистрофия Эмери − Дрейфуса;
- скапулоперонеальная;
- конгенитальная (несколько доброкачественных заболеваний, выявляющихся сразу после рождения или в раннем детском возрасте);
- лице-лопаточно-плечевая миопатия Ландузи − Дежерина;
- окулофарингеальная (поражающая глоточные и глазодвигательные мышцы);
- лопаточно-перонеальная миопатия Давиденкова;
- дистальная мышечная дистрофия.
В основу наследственных форм амиотрофий положены генетические нарушения синтеза мышечных белков или отвечающих за регуляцию обмена веществ ферментов.
Приобретенные миопатии:
- мембранные – возникающие как следствие дефектов в мембранных системах мышечных волокон;
- воспалительные – появляются на фоне инфекционно-воспалительных (бактериальных, вирусных, паразитарных) поражений мышц;
- токсические – итог хронических интоксикаций (профессиональной, алкогольной, наркотической и пр.);
- метаболические – связаны с расстройством обмена липидов, гликогена, нарушением метаболизма пуринов или работы митохондрий.
Симптомы миопатии
Большинство миопатий развивается постепенно. На ранних стадиях патологического процесса пациенты жалуются на мышечную слабость в руках и ногах и усталость, возникающую после ходьбы и незначительной физической нагрузки.
По мере прогрессирования заболевания миастения усиливается, мышцы начинают истончаться и уменьшаться в размерах, формируются деформации конечностей. У больных возникают существенные трудности при подъеме и спуске по лестнице, поднятии с пола и даже со стула, они начинают передвигаться, раскачиваясь из стороны в сторону.
Из-за чрезмерного изгиба позвоночника в области поясницы живот выпячивается вперед, плечи опускаются, лопатки приобретают крыловидную форму.
Клиническая картина
При миопатиях чаще всего поражаются проксимальные отделы верхних и нижних конечностей, из-за чего дистальные приобретают гипертрофированный вид. При этом происходят симметричные атрофии мышечных групп. Вместе с нарастанием мускульной слабости угасают сухожильные рефлексы, что, в свою очередь, приводит к суставным контрактурам и вялому периферическому параличу.
При некоторых формах миопатий поражается диафрагмальная мускулатура. Это влечет за собой развитие застойной пневмонии и дыхательной недостаточности. Нередко выявляются патологические изменения со стороны сердечно-сосудистой системы. Поражение мышц гортаноглотки становится причиной дисфагии (расстройства глотания) и миопатического пареза (двигательных нарушений) гортани.
Лечение миопатии
На сегодняшний день первичная миопатия лечится только симптоматически. Методика, направленная на улучшение метаболических процессов в мышечной ткани, включает в себя комплексную медикаментозную терапию, ЛФК, плавание и ортопедическую коррекцию при помощи специальных протезов.
Лечение вторичных форм миопатии направлено на устранение основного заболевания и дезинтоксикацию организма.
Прогрессирующие мышечные дистрофии
Прогрессирующие мышечные дистрофии (ПМД) — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
КЛИНИЧЕСКАЯ КАРТИНА
Для всех ПМД типичны мышечная слабость различной степени выраженности и мышечные атрофии. Тип распределения мышечной слабости при ПМД — один из основных диагностических критериев. Для каждой из форм ПМД характерно избирательное поражение определённых мышц при сохранности других, рядом расположенных.
В целом типичный миопатический симптомокомплекс включает следующие признаки.
• Симметричную проксимальную мышечную слабость различной степени выраженности (мышечная сила от 3-4 баллов на ранней и до 1-0 — на поздних стадиях заболевания) , постепенно развивающиеся атрофии мышц.
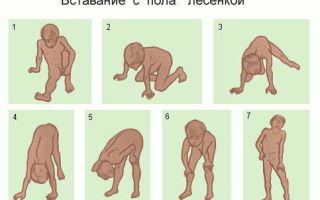
• Симптом Говерса: больной, для того чтобы подняться из положения на корточках, опирается руками об пол, затем поднимается, опираясь руками об колени, — «взбирается по себе». Этот рано появляющийся симптом обусловлен слабостью мышц бёдер и тазового пояса.
• Затруднения при ходьбе по лестнице — больной помогает себе с помощью рук.
• «Утиную» (переваливающуюся) походку, связанную со слабостью мышц тазового пояса.
• Поясничный гиперлордоз, обусловленный слабостью мышц тазового пояса и спины.
• «Крыловидные» лопатки вследствие слабости передней зубчатой мышцы, а также других мышц, фиксирующих лопатку.
• Псевдогипертрофию икроножных мышц вследствие развития в них соединительной ткани (сила мышц при этом снижена) .
• Ходьбу на цыпочках из-за контрактур ахилловых сухожилий.
• Сохранность экстраокулярных мышц, мышц лица.
Миопатический симптомокоплекс наиболее отчётливо выявляют при ПМД Дюшенна и Беккера.
• Для ПМД Дюшенна характерно раннее начало заболевания (в 3-7 лет) , быстрое прогрессирование, высокие показатели КФК, выраженная спонтанная активность по данным игольчатой ЭМГ, отсутствие дистрофина в мышцах при иммуногистохимическом исследовании.
По мере прогрессирования мышечной слабости затрудняется самостоятельная ходьба и уже в 9-15 лет больные вынуждены пользоваться инвалидным креслом, что провоцирует развитие кифосколиоза, остеопороза. На поздних стадиях у большинства больных развиваются дилатационная кардиомиопатия, слабость дыхательной мускулатуры. Интеллект чаще всего умеренно снижен.
• Клинические про явления ПМД Беккера в целом напоминают таковые при форме Дюшенна, но течение заболевания более мягкое: дебют приходится на более поздний возраст (от 2 до 21 года, в среднем в 11 лет) , летальный исход наступает позже (в 23-63 года) .
• Конечностно-поясные формы ПМД также характеризуются развитием миопатического симптомокомплекса.
ПМД Эрба по возрасту начала заболевания, скорости прогрессирования и клиническим проявлениям напоминает форму Беккера, однако для неё не характерна кардиальная патология, кроме того, заболевание отмечают как у мальчиков, так и девочек. При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия.
• ПМД Ландузи-Дежерина характеризуется выраженной слабостью мимических мышц (за исключением редкой формы без мимической слабости), симптомом «крыловидных» лопаток, слабостью дву- и трёхглавых мышц плеча при интактных дельтовидных мышцах, степпажем.
Как правило, интактными остаются экстраокулярные мышцы (за исключением одного подтипа) и мышцы языка и глотки, дыхательная мускулатура. У некоторых больных возникает слабость мышц тазового пояса (около 20% больных вынуждены пользоваться инвалидным креслом) . Мышечные атрофии часто бывают асимметричными.
У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
• ПМД Эмери-Дрейфуса характеризуется наличием контрактур (чаще в локтевых, коленных суставах, задних мышцах шеи, из-за которых голова оказывается слегка запрокинутой) и плечелопаточно-перонеальным распределением мышечной слабости и атрофий с сохранностью лицевой мускулатуры. Часто отмечают нарушения ритма сердца и кардиомиопатию. Заболевание часто дебютирует с контрактур.
• Основной симптом офтальмофарингеальной формы — хроническая прогрессирующая наружная офтальмоплегия, затем присоединяется умеренный бульбарный синдром. В дальнейшем развивается проксимальная мышечная слабость в руках и ногах.
• Дистальные миопатии характеризуются преобладанием слабости дистальных мышц. При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, при миопатии Миоши — икроножные мышцы: больные плохо стоят на носках, часто спотыкаются.
При миопатии Говерса, тибиальной миопатии главный симптом — степпаж из-за слабости перонеальной группы мышц, при этом миопатия Говерса склонна к дальнейшей генерализации: через 5-10 лет присоединяется слабость кистей и мышц шеи, часто отмечают «свисание» 1 пальца на ногах и V — на руках.
При тибиальной миопатии, распространённой в Финляндии, чаще всего наблюдают изолированное поражение передних больше берцовых мышц, иногда развивается кардиомиопатия.
СИМПТОМЫ
При ПМД Дюшенна, Беккера, конечностно-поясных формах проявляется наиболее выраженная слабость в пояснично-подвздошных мышцах, мышцах бёдер, дельтовидных, дву- и трёхглавых мышцах плеча.
Менее выражена слабость в дистальных мышцах конечностей. Лицевые мышцы остаются сохранными. Наряду с мышечной слабостью постепенно развиваются гипотрофии поражённых мышц вплоть до атрофии на поздних стадиях.
При этом соседние мышцы могут быть полностью клинически интактны.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
Немедикаментозное лечение
Чрезмерная физическая нагрузка, как и недостаточная, приводит к нарастанию мышечной слабости. Ежедневная ЛФК позволяет поддерживать мышечный тонус и препятствует развитию контрактур.
Комплекс ЛФК обязательно должен включать активные и пассивные упражнения, упражнения на растяжку/предупреждение контрактур и дыхательную гимнастику. Активный массаж с разминанием мышц может усиливать мышечную слабость и утомляемость, поэтому рекомендуют щадящий массаж.
Физиотерапевтическое лечение больные переносят по-разному: некоторые не ощущают улучшений или даже жалуются на усиление мышечной слабости.
Хирургическое лечение
В некоторых случаях возможно хирургическое лечение контрактур, однако при этом необходимо помнить о возможности увеличения мышечной слабости за время восстановительного лечения (вплоть до потери способности к ходьбе). В ряде случаев необходима имплантация кардиостимулятора.