Талассемия – наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
Талассемия – группа генетически детерминированных болезней крови, развивающихся при нарушении синтеза a- или β-цепей гемоглобина, сопровождающихся гемолизом, гипохромной анемией, микроцитозом. В гематологии талассемия относится к наследственным гемолитическим анемиям — количественным гемоглобинопатиям.
Талассемия широко распространена среди населения Средиземноморского и Черноморского региона; название заболевания буквально переводится как «анемия морского побережья».
Также случаи талассемии нередки в странах Африки, Ближнего Востока, Индии и Индонезии, Средней Азии и Закавказья. С синдромом талассемии каждый год в мире рождается 300 тыс. детей.
В зависимости от формы патологии течение талассемии может быть тяжелым, фатальным или легким, бессимптомным. Так же, как серповидно-клеточная анемия, талассемия играет роль защитного фактора против малярии.
Талассемия
Талассемия является генетическим заболеванием с аутосомно-рецессивным наследованием. Непосредственной причиной патологии выступают различные мутационные нарушения в гене, кодирующем синтез той или иной цепи гемоглобина.
Молекулярную основу дефекта могут составлять синтез аномальной матричной РНК, делеции структурных генов, мутации регуляторных генов либо их неэффективная транскрипция.
Следствием подобных нарушений служит снижение или отсутствие синтеза одной из полипептидных гемоглобиновых цепей.
Так, при b-талассемии бета-цепи синтезируются в недостаточном количестве, что приводит к избытку альфа-цепей, и наоборот. Избыточно продуцируемые полипептидные цепи откладываются в клетках эритроидного ряда, вызывая их повреждение.
Это сопровождается деструкцией эритрокариоцитов в костном мозге, гемолизом эритроцитов в периферической крови, гибелью ретикулоцитов в селезенке. Кроме этого, при b-талассемии в эритроцитах накапливается фетальный гемоглобин (НbF), не способный транспортировать кислород в ткани, что вызывает развитие тканевой гипоксии.
Вследствие костномозговой гиперплазии развивается деформация костей скелета. Анемия, тканевая гипоксия и неэффективный эритропоэз в той или иной степени нарушают развитие и рост ребенка.
Для гомозиготной формы талассемии характерно наличие двух дефектных генов, унаследованных от обоих родителей. При гетерозиготном варианте талассемии пациент является носителем мутантного гена, унаследованного от одного из родителей.
С учетом поражения той или иной полипептидной цепи гемоглобина различают:
- a-талассемию (с подавлением синтеза альфа-цепей HbA). Данная форма может быть представлена гетерозиготным носительством манифестного (α-th1) или немого (α-th2) гена; гомозиготной a-талассемией (водянкой плода с гемоглобином Бартса); гемоглобинопатией Н
- b-талассемию (с подавлением синтеза бета-цепей HbA). Включает в себя гетерозиготную и гомозиготную β-талассемию (анемию Кули), гетерозиготную и гомозиготную δβ-талассемию (F-талассемию)
- γ-талассемию (с подавлением синтеза гамма-цепей гемоглобина)
- δ-талассемию (с подавлением синтеза дельта-цепей гемоглобина)
- талассемию, обусловленную нарушением структуры гемоглобина.
Статистически чаще встречается β-талассемия, которая, в свою очередь, может протекать в 3-х клинические формах: малой, большой и промежуточной. По тяжести синдрома выделяют легкую форму талассемии (пациенты доживают до половой зрелости), средне-тяжелую (продолжительность жизни больных 8-10 лет) и тяжелую (гибель детей наступает в первые 2-3 года жизни).
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов.
Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей.
Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, — сахарного диабета; кардиосклероза и сердечной недостаточности.
Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется.
Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности.
Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Талассемия
Талассемию следует заподозрить у лиц с семейным анамнезом, характерными клиническими признаками и лабораторными показателями. Больные талассемией нуждаются в консультации гематолога и медицинского генетика.
Типичными гематологическими изменениями служат снижение уровня гемоглобина и цветового показателя, гипохромия, наличие мишеневидных эритроцитов, повышение уровня железа сыворотки крови и непрямого билирубина.
Электрофорез Hb на ацетат-целлюлозной пленке используется для определения различных гемоглобиновых фракций. При изучении пунктата костного мозга обращает внимание гиперплазия красного кроветворного ростка с высоким числом эритробластов и нормобластов.
Молекулярно-генетические исследования позволяют выявить мутацию в локусе a- или β-глобина, нарушающую синтез полипептидной цепи.
На краниограммах при большой b-талассемии выявляется игольчатый периостоз (феномен «волосатого черепа»). Характерна поперечная исчерченность трубчатых и плоских костей, наличие мелких очагов остеопороза. С помощью УЗИ брюшной полости обнаруживается гепатоспленомегалия, камни желчного пузыря.
При подозрении на талассемию требуется исключить железодефицитную анемию, наследственный микросфероцитоз, серповидно-клеточную анемию, аутоиммунную гемолитическую анемию.
В семьях, имеющих больных талассемией, рекомендуется проведение генетического консультирования супругов и инвазивной дородовой диагностики (биопсии хориона, кордоцентеза, амниоцентеза) для выявления гемоглобинопатии на ранних сроках беременности.
Подтверждение гомозиготных форм талассемии у плода служит показанием для искусственного прерывания беременности.
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются.
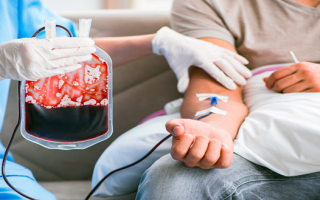
С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Талассемия 1
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте.
При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают.
Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства – отказ от деторождения.
Талассемия — Причины, Симптомы, Диагностика и Лечение | Спиженко
Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.
Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
- Если изменен один ген цепи – заболевание (малая талассемия), протекает легко и бессимптомно. Но, при этом человек остается носителем данной болезни, которую может передать по наследству своим детям.
- Нарушение синтеза 2-х генов проявляется микроцитарной анемией в легкой степени тяжести.
- Патология 3-х генов протекает тяжело с ярко выраженной симптоматикой.
- Повреждение сразу 4-х генов (большая талассемия) – это наиболее редкий вид талассемии, который практически несовместим с жизнью (иногда ситуация решается пересадкой костного мозга).
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
- Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
- Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
- Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
- Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Причины
Главная причина талассемии – наследственный фактор. При котором у одного пациента функционирует патологическая РНК (участвует в кодировании, чтении и регуляции генов), а у другого – ДНК (появляются изменения в хромосоме). На фоне этих изменений уменьшается или полностью прекращается синтез одной из цепочек Hb.
В норме количество альфа, бета-цепей – одинаково, поэтому изменение синтеза хотя бы одной из них приводит к дисбалансу и патологии.
Болезнь встречается как у девочек, так и у мальчиков. Частота появления большой талассемии 1:100 000, если оба родители носители мутационного гена.
Также на патогенез талассемии влияет и этническая принадлежность, так как чаще всего данная генетическая патология встречается у жителей Африки, Восточного Средиземноморья и Юго-Восточной, Центральной Азии.
Определено это тем, что мутационный ген имеет большую сопротивляемость к малярии (обычно в африканских регионах) и большим количеством родственных браков, что приводит к разным генетическим нарушениям, включая талассемию.
В Украине талассемия – редкая патология.
Всемирная организация здравоохранения приняла международную классификацию заболеваний 10-го пересмотра (мкб-10), как единый нормативный документ для ведения учета болезней, причин обращений пациентов в медицинские учреждения и причин смертности.
Согласно мкб талассемии (D56) имеет следующую классификацию:
- D56.0 – альфа-талассемия;
- D56.1 – бета-талассемия;
- D56.2 – дельта-бета талассемия;
- D56.3 – носитель талассемии;
- D56.9 – талассемия неуточненная.
Обновление кодов (мкб-11) будет вводиться в действие с 2022 года.
Симптомы
Талассемия определяется до рождения ребенка при помощи пренатального скрининга.
Если на данном этапе болезнь не была выявлена, следует ожидать такие симптомы:
- Деформация костей черепа;
- Увеличение живота;
- Желтушность/бледность кожи и слизистых оболочек.
По результатам лабораторных анализов:
- Снижение уровня гемоглобина в крови и цветового показателя;
- Наличие эритроцитов неправильной формы;
- Повышение билирубина и показателей железа;
- Повышение уровня эритробластов, нормобластов.
По результатам УЗИ диагностики:
- Увеличение селезенки (спленомегалия);
- И печени.
Проявление первых симптомов талассемии зависит от вида и количества «деформированных» генов.
Симптомы большой альфа-талассемии проявляются сразу после рождения, большой бета-талассемии – в первые два года жизни малыша.
Поэтому, бета-талассемия может быть дополнена следующими признаками:
- Замедленным ростом;
- Наличием темной мочи;
- Повышенной усталостью (гиподинамия);
- Наличием железодефицитной анемии;
- Одышкой;
- Симптомами интоксикации.
Включая симптомы сердечно-сосудистых, эндокринных патологий и заболеваний печени.
При талассемии резко увеличивается усвоение железа из продуктов питания – это приводит к дисбалансу работы внутренних органов и систем. При отсутствии корректной диагностики и лечения талассемии – остается высокий риск летального исхода в первый год жизни.
Основные нарушения в организме, к которым приводит талассемия:
- Анемия.
- Патологическое накопление железа в крови.
- Нарушение работы сердечной мышцы.
- Поджелудочной железы, гипофиза.
- Фиброза печени (далее цирроза).
- Нарушение полового созревания (гипогонадизм).
- Спленомегалия (увеличение селезенки).
- Расширение участков кроветворения (в костном мозге).
Если у пациента поражен только один ген – талассемия не имеет симптоматических проявлений.
Однако следить за уровнем гемоглобина и размером селезенки – необходимо.
Диагностика
Так как симптоматика талассемии не всегда бывает специфической — врач для постановки окончательного диагноза, в большей мере опирается на результаты лабораторных исследований крови и спинномозговой жидкости.
Поэтому, при подозрении на талассемию необходимо:
- Сдать общий анализ крови.
- Биохимический анализ крови.
- Пройти ПЦР (молекулярные тесты).
- УЗИ диагностику.
- Рентгенографию.
Во время исследования крови обращают внимание:
- На количество, форму, размер эритроцитов;
- Уровень гемоглобина, билирубина;
- Цветовой показатель крови;
- Соотношение HbA и HbF;
- насыщение трансферрина железом;
- Количество ретикулоцитов;
- Концентрацию ферритина в сыворотке крови и т.д.
При исследовании костного мозга:
- Наличие/отсутствие эритроидной гиперплазии.
УЗИ и рентгенография нацелены:
- На определение размеров внутренних органов и желез (селезенки, печени, щитовидки);
- Изучение кортикального слоя костей черепа, наличие/отсутствие гранул, расширений, уплотнений в трубчатых, губчатых костях, позвонках и т.д.
Для того чтобы предотвратить негативные последствия болезни, родителям рекомендуется в профилактических целях в период беременности пройти внутриутробный скрининг плода на определение разных генетических патологий, включая талассемию.
Существуют два способа пренатальной диагностики:
- Биопсия ворсинок хориона (на 11-й неделе беременности).
- Отбор околоплодных вод (на 16-й неделе беременности).
Что позволит своевременно определить заболевание и предпринять все необходимые меры.
Лечени
Малая талассемия (нарушение одного гена) – не требует лечения. Достаточно здорового образа жизни, сбалансированного питания, физической активности и отсутствия пагубных привычек. Все это «поможет» организму самостоятельно компенсировать дефект гена.
Большая талассемия и средней степени тяжести нуждается в специальном лечении, которое в «Клинике Спиженко» включает:
- Трансфузионную терапию (переливание эритроцитарной массы);
- Хелатную терапию (выведение лишнего железа из организма);
- Спленэктомию (удаление селезенки);
Лекарственные препараты при талассемии назначаются только для коррекции симптомов и предупреждения осложнений. Медикаментозного лечения талассемии – нет.
Пациенты с талассемией средней тяжести в донорских трансфузиях нуждаются не часто, только в критических ситуациях для организма:
- При беременности;
- Сердечной недостаточности;
- Резком снижении уровня гемоглобина (меньше 50 г/л);
- Оперативном вмешательстве;
- Заболеваниях опорно-двигательного аппарата;
- Легочной гипертензии.
Большая талассемия требует более серьезного лечения:
- Трансфузия (1 раз в месяц на протяжении всей жизни);
- Удаление избытка железа (следствие частых трансфузий);
- Удаление селезенки (при спленомегалии);
- Трансплантация стволовых клеток.
Удаление селезенки уменьшает потребность в гемотрансфузиях. Однако отсутствие природного «фильтра» увеличивает риск возникновения разнообразных инфекций, что может негативно отразиться на самочувствии пациента.
- Единственным, но радикальным вариантом лечения тяжелых пациентов с талассемией, остается пересадка костного мозга.
- Данная терапия позволит и детям и взрослым с талассемией вести нормальный образ жизни с максимальным сохранением ее качества.
- Отсутствие должного лечения большой бета-талассемии приводит к ранней смертности пациентов
- Для контроля результатов лечения талассемии назначается:
- Общий анализ крови;
- Биохимический (с оценкой работы почек и печени);
- УЗИ;
- ЭХО-ЭКГ.
Включая изучение гормонального фона (для пациентов с 8 лет), минеральной плотности костей, остроты зрения и слуха.
Правильный образ жизни и серьезный подход к лечению при талассемии средней тяжести не представляет собой угрозы для нормальной жизнедеятельности пациента. Поэтому прогноз протекания болезни остается достаточно оптимистичным.
Чего нельзя сказать о тяжелой талассемии. Без должного комплексного лечения уменьшается не только продолжительность жизни, но и ее качество.
Поэтому, тем людям, у которых в семье есть родственники с данным наследственным отклонением, нужно пройти комплексное генетическое исследование в обязательном порядке. Особенно в том случае, если вы планируете рождение ребенка.
Талассемия
Талассемия — это наследственное заболевание крови, характеризующееся пониженной выработкой гемоглобина . [7] Симптомы зависят от типа и могут варьироваться от отсутствия до тяжелых.
[1] Часто бывает анемия от легкой до тяжелой (низкий уровень эритроцитов или гемоглобина). [1] Анемия может привести к чувству усталости и бледности кожи . [1] Также могут быть проблемы с костями, увеличенная селезенка , желтоватая кожа и темная моча.
[1] У детей может наблюдаться медленный рост. [1]
Талассемия — это генетическое заболевание, унаследованное от родителей. [2] Существует два основных типа: альфа-талассемия и бета-талассемия .
[7] Тяжесть альфа- и бета-талассемии зависит от того, сколько из четырех генов альфа-глобина или двух генов бета-глобина отсутствует.
[2] Диагноз обычно ставится на основании анализов крови, включая общий анализ крови , специальные тесты на гемоглобин и генетические тесты. [3] Диагноз может быть поставлен до рождения с помощью пренатального тестирования . [8]
Лечение зависит от типа и степени тяжести. [4] Лечение более тяжелых заболеваний часто включает регулярное переливание крови , хелатирование железа и фолиевую кислоту . [4] Хелатирование железа может быть выполнено с помощью дефероксамина , деферасирокса или деферипрона .
[4] [9] Иногда возможна трансплантация костного мозга . [4] Осложнения могут включать перегрузку железом в результате переливания крови, что может привести к заболеваниям сердца или печени , инфекциям и остеопорозу . [1] Если селезенка чрезмерно увеличена, может потребоваться хирургическое удаление .
[1] Пациенты с талассемией, которые плохо реагируют на переливание крови, могут принимать гидроксимочевину или талидомид , а иногда и их комбинацию. [10] Гидроксимочевина — единственный одобренный FDA препарат для лечения талассемии.
У пациентов, которые принимали 10 мг / кг гидроксимочевины каждый день в течение года, уровень гемоглобина был значительно выше, и это было хорошо переносимым лечением для пациентов, которые плохо реагировали на переливание крови.
[11] Другой индуктор гемоглобина включает талидомид, хотя он не был протестирован в клинических условиях. Комбинация талидомида и гидроксимочевины приводила к значительному повышению уровня гемоглобина у пациентов, зависимых от трансфузий и не зависимых от трансфузий [12]
По состоянию на 2015 год талассемия встречается примерно у 280 миллионов человек, из которых около 439000 страдают тяжелыми заболеваниями. [13] Это наиболее распространено среди людей итальянского, греческого, турецкого, ближневосточного , южноазиатского и африканского происхождения. [7] У мужчин и женщин одинаковые показатели заболеваемости.
[14] Это привело к 16 800 смертельным случаям в 2015 году по сравнению с 36 000 смертей в 1990 году. [6] [15] Те, у кого есть незначительная степень талассемии, аналогичная тем, у кого есть серповидноклеточная черта , имеют некоторую защиту от малярии , что объясняет, почему они чаще встречаются в регионах мира, где существует малярия.
[16]
Слева: рука человека с тяжелой анемией. Справа: рука человека без анемии.
- Перегрузка железом : люди с талассемией могут получить перегрузку железом в организме либо из-за самого заболевания, либо из-за частых переливаний крови. Слишком много железа может привести к повреждению сердца, печени и эндокринной системы , в том числе желез, вырабатывающих гормоны, регулирующие процессы во всем организме. Повреждение характеризуется чрезмерным отложением железа. Без адекватной терапии хелатированием железа почти у всех пациентов с бета-талассемией накапливается потенциально смертельный уровень железа. [17]
- Инфекция: люди с талассемией имеют повышенный риск заражения. Это особенно актуально, если селезенка была удалена. [18]
- Деформации костей: талассемия может вызвать расширение костного мозга, что приводит к расширению костей. Это может привести к аномальной структуре костей, особенно на лице и черепе. Расширение костного мозга также делает кости тонкими и ломкими, что увеличивает риск переломов костей. [19]
- Увеличенная селезенка : селезенка помогает бороться с инфекцией и фильтрует нежелательные вещества, такие как старые или поврежденные клетки крови. Талассемия часто сопровождается разрушением большого количества эритроцитов, и задача удаления этих клеток приводит к увеличению селезенки. Спленомегалия может усугубить анемию и сократить срок жизни перелитых эритроцитов. Сильное увеличение селезенки может потребовать ее удаления. [20]
- Замедление темпов роста: анемия может вызвать замедление роста ребенка. Половое созревание также может задерживаться у детей с талассемией. [21]
- Проблемы с сердцем: такие заболевания, как застойная сердечная недостаточность и нарушение сердечного ритма, могут быть связаны с тяжелой талассемией. [22]
Талассемия имеет
аутосомно-рецессивный тип наследования.
Как α-, так и β-талассемии часто наследуются по аутосомно- рецессивному типу. Сообщалось о случаях доминантно наследуемой α- и β-талассемии, первая из которых была в ирландской семье с двумя делециями 4 и 11 п.н.
в экзоне 3, прерванных вставкой 5 п.н. в ген β-глобина. Для аутосомно-рецессивных форм заболевания оба родителя должны быть носителями, чтобы ребенок был поражен.
Если оба родителя несут признак гемоглобинопатии, риск каждой беременности для пораженного ребенка составляет 25%.
Эволюция
Наличие единственного генетического варианта талассемии может защитить от малярии и, следовательно, может быть преимуществом. [23]
Люди, у которых диагностирована гетерозиготная (носительская) β-талассемия, имеют некоторую защиту от ишемической болезни сердца . [24]
Обычно большая часть взрослого гемоглобина ( HbA ) состоит из четырех белковых цепей, двух α и двух β-глобиновых цепей, собранных в гетеротетрамер .
При талассемии пациенты имеют дефекты в цепи α- или β-глобина, вызывающие выработку аномальных эритроцитов (при серповидно-клеточной анемии , которая представляет собой гемоглобинопатию, а не правильную талассемию, мутация специфична для β-глобина).
Талассемии классифицируются в зависимости от того, какая цепь молекулы гемоглобина поражена. При α-талассемии нарушается продукция цепи α-глобина, тогда как при β-талассемии нарушается продукция цепи β-глобина.
Цепи β-глобина кодируются одним геном на хромосоме 11 ; Цепи α-глобина кодируются двумя тесно связанными генами на хромосоме 16 .
[25] Таким образом, у нормального человека с двумя копиями каждой хромосомы два локуса кодируют β-цепь, а четыре локуса кодируют α-цепь.
Делеция одного из α-локусов широко распространена у лиц африканского или азиатского происхождения, что повышает вероятность развития α-талассемии . β-Талассемии распространены не только у африканцев , но также у греков , турок и итальянцев .
Альфа-талассемии
В α-талассемии вовлечены гены HBA1 [26] и HBA2 , [27], унаследованные рецессивно по менделевской модели . Существуют два генных локуса и, следовательно, четыре аллеля. Для α-глобина существует два генетических локуса, таким образом, четыре аллеля находятся в диплоидных клетках.
Два аллеля являются материнскими, а два — отцовскими. Тяжесть α-талассемии коррелирует с количеством пораженного α-глобина; аллели: чем больше, тем тяжелее будут проявления болезни.
[28] Альфа-талассемия приводит к снижению выработки альфа-глобина; следовательно, образуется меньше цепей альфа-глобина, что приводит к избытку β-цепей у взрослых и избытку γ-цепей у новорожденных.
Избыточные β-цепи образуют нестабильные тетрамеры (называемые гемоглобином H или HbH 4 β-цепей), которые имеют аномальные кривые диссоциации кислорода. Альфа-талассемия часто встречается у людей из Юго-Восточной Азии, Ближнего Востока, Китая и лиц африканского происхождения. [29]
| 1 | Бесшумный носитель | Нет симптомов |
| 2 | Признак альфа-талассемии | Незначительная анемия |
| 3 | Болезнь гемоглобина H | Легкая или умеренная анемия; может вести нормальную жизнь |
| 4 | Водянка плода | Смерть плода обычно наступает при рождении. |
Бета-талассемия
Бета-талассемии возникают из-за мутаций в гене HBB на хромосоме 11 [30], также наследуются аутосомно, рецессивно. Тяжесть заболевания зависит от характера мутации и от наличия мутации в одном или обоих аллелях.
Мутировавшие аллели называются β +, когда частичная функция сохраняется (либо белок имеет пониженную функцию, либо он функционирует нормально, но вырабатывается в пониженном количестве), или β o , когда функционирующий белок не продуцируется.
Положение обоих аллелей определяет клиническую картину:
- Большая β-талассемия ( средиземноморская анемия или анемия Кули ) вызывается генотипом β o / β o . Функциональные β-цепи не образуются, поэтому сборка гемоглобина А невозможна. Это самая тяжелая форма β-талассемии;
- Промежуточная β-талассемия вызывается генотипом β + / β o или β + / β + . В этой форме вырабатывается некоторое количество гемоглобина А;
- Малая β-талассемия вызывается генотипом β / β o или β / β + . Только один из двух аллелей β-глобина содержит мутацию, поэтому образование β-цепей не сильно нарушено, и пациенты могут быть относительно бессимптомными.
Бета-талассемия чаще всего встречается у людей средиземноморского происхождения. В меньшей степени могут пострадать китайцы, выходцы из Азии и афроамериканцы. [29]
Дельта-талассемия
Помимо альфа- и бета-цепей, присутствующих в гемоглобине, около 3% взрослого гемоглобина состоит из альфа- и дельта-цепей. Как и в случае с бета-талассемией, могут возникать мутации, влияющие на способность этого гена продуцировать дельта-цепи. [ необходима цитата ]
Комбинированные гемоглобинопатии
Талассемия может сосуществовать с другими гемоглобинопатиями . Самые распространенные из них:
Талассемию можно диагностировать с помощью общего анализа крови , электрофореза гемоглобина и анализа ДНК.
[32] Электрофорез гемоглобина не является широко доступным в развивающихся странах, поэтому индекс Ментцера также может использоваться для диагностики талассемии. Хотя это не диагностический тест, но он может дать четкое представление о возможности талассемии.
Индекс Ментцера можно рассчитать на основании отчета об общем анализе крови. Для этого также можно использовать калькуляторы индекса Ментцера. [33]
Американский колледж акушеров и гинекологов рекомендует всем людям думающие забеременеть быть проверена , чтобы увидеть , если они имеют талассемии. [34] Семьям с признаками талассемии рекомендуется генетическое консультирование и генетическое тестирование .
На Кипре существует политика скрининга, направленная на снижение заболеваемости талассемией, которая с момента реализации программы в 1970-х годах (которая также включает пренатальный скрининг и аборты) снизила количество детей, рожденных с этим заболеванием, с одного из 158 рождений почти до нуль. [35] В Греции также действует программа скрининга для выявления носителей. [36]
В Иране в качестве добрачного скрининга сначала проверяют показатели эритроцитов мужчины. Если у него есть микроцитоз ( средний гемоглобин клеток
Талассемии | благотворительный фонд «Подари жизнь»
Талассемии – группа наследственных заболеваний кроветворной системы, которые характеризуются нарушением синтеза гемоглобина. Поэтому основным проявлением болезни является анемия.
Как известно, нормальный человеческий гемоглобин HbA состоит из четырех белковых цепей двух разных видов: две α-цепи и две β-цепи.
Соответственно, если нарушен синтез α-цепей (при этом в крови появляется аномальный гемоглобин, состоящий из четырех β-цепей), то говорят об альфа-талассемии.
Если же нарушен синтез β-цепей, то речь идет о бета-талассемии; при этом образуются другие варианты гемоглобина, которые не содержат β-цепей. Чаще встречается именно бета-талассемия. Существуют также другие варианты болезни, но их клиническая значимость невелика.
Перечисленные разновидности болезни подразделяются еще на несколько подтипов в зависимости от числа и вида генетических дефектов. Так, у бета-талассемии выделяют большую, промежуточную и малую формы; наиболее тяжелым заболеванием является большая талассемия, тогда как малая форма бета-талассемии позволяет вести практически нормальную жизнь.
Частота встречаемости, факторы риска
Талассемии – наследственные заболевания.
Тяжелые формы талассемии наследуются по аутосомно-рецессивному механизму, то есть больные дети могут родиться в тех семьях, где и отец, и мать являются носителями дефектного гена – однако соответствующие аномалии крови у родителей выражены слабо, так как у них есть и «здоровые» копии необходимого гена. Так, у двух родителей с малой формой бета-талассемии может родиться ребенок с большой талассемией.
Болезнь встречается как у мальчиков, так и у девочек. Частота ее составляет около 1 на 100 тысяч в среднем по всему миру, но зависит прежде всего от региона. Большинство больных талассемией – жители южных стран (Средиземноморье, страны Центральной и Южной Азии).
Это связано с тем, что носительство «дефектного» гена ведет к лучшей сопротивляемости заболеванию малярией. Поэтому в регионах с широким распространением малярии число носителей и, соответственно, число больных талассемией может быть довольно велико.
В России больных большой бета-талассемией сравнительно мало, при этом многие из них – потомки жителей Поволжья, Кавказа или Средней Азии.
Семьям, где уже были случаи рождения детей с тяжелыми формами талассемии, рекомендуется консультация генетика. Так как носительство «дефектного» гена при бета-талассемии легко определяется по результатам простых и недорогих анализов крови, в странах с широким распространением талассемии развиваются программы профилактики этого заболевания.
Признаки и симптомы
При талассемии наблюдаются обычные для анемии симптомы: бледность, одышка, плохая переносимость физических нагрузок, сниженный аппетит. При большой форме бета-талассемии анемия обычно начинает проявляться в возрасте нескольких месяцев.
У детей нередко встречается задержка роста. Могут возникать боли в животе из-за увеличения селезенки и камней в желчном пузыре. Нередко наблюдаются увеличение печени, желтушность кожи и слизистых оболочек, деформации костей (из-за нарушения функционирования костного мозга), аномалии прикуса, сердечная недостаточность и/или аритмии и др.
Диагностика
При талассемии наблюдаются характерные изменения в клиническом анализе крови. Резко снижены количество эритроцитов, уровень гемоглобина (при тяжелых формах – иногда до 20–30 г/л) и цветовой показатель.
Эритроциты крови уменьшенного размера, зачастую необычного вида – так называемые «мишеневидные».
Для диагностики полезно использовать также результаты биохимического исследования крови и, возможно, анализ образца костного мозга.
Анализ гемоглобина методом электрофореза, а также биохимическое определение фетального гемоглобина позволяют подтвердить диагноз. При талассемиях снижено количество «нормального» гемоглобина, но повышено количество аномального.
Полезен сбор семейного анамнеза для установления наследственной природы анемии. В отдельных случаях может проводиться генетический анализ для выявления конкретного генетического дефекта.
Лечение
Для лечения анемии при большой форме талассемии применяются переливания эритроцитарной массы – например, раз в месяц или два месяца. Они позволяют поддерживать приемлемый уровень гемоглобина.
Однако регулярные переливания приводят к накоплению в организме избытка железа – то есть перегрузке железом.
Постепенно этот избыток становится опасным для жизни, и больные должны принимать специальные лекарства-хелаторы для выведения железа из организма.
Если сильно увеличена селезенка (спленомегалия), может быть рекомендовано ее хирургическое удаление. Увеличение селезенки при талассемии связано как с ее повышенным участием в разрушении аномальных эритроцитов, так и с происходящими в ней процессами неэффективного кроветворения.
Удаление селезенки приводит к некоторому улучшению состояния, но повышает риск инфекций и других осложнений.
В последние годы появилась информация, что части пациентов может помочь терапия препаратом «Джакави» (руксолитиниб): исследования показывают, что такая терапия в случае успеха приводит к уменьшению размеров селезенки и снижению потребности в переливаниях, а также, в сочетании с иммуносупрессивными препаратами, повышает шансы на успех последующей трансплантации.
Так как речь идет о генетически обусловленном заболевании, никакая консервативная терапия не может привести к полному излечению. Единственный шанс на нормализацию кроветворения дает аллогенная трансплантация костного мозга. Однако это сложная процедура, связанная с риском для жизни.
При наличии совместимого здорового донора среди братьев и сестер пациента трансплантация обычно проходит успешно. Трансплантации от других доноров сейчас проводятся все чаще, но они бывают сопряжены с серьезными трудностями, прежде всего с повышенным риском отторжения.
Поэтому врачи все время разрабатывают новые протоколы подготовки к трансплантации.
Прогноз
Раньше тяжелые формы талассемии (такие как большая форма бета-талассемии) обычно приводили к смерти в раннем детском возрасте. Ситуация изменилась в последние десятилетия.
Сейчас периодические переливания крови в сочетании с терапией, направленной на удаление избытка железа, часто позволяют больным доживать до среднего и даже пожилого возраста.
Однако следует помнить, что эта терапия не ведет к полному излечению и должна быть пожизненной.
Трансплантация костного мозга в случае успеха приводит к нормализации кроветворения, но ее проведение связано с определенными рисками, особенно есть у больного нет совместимого родственного донора.
Талассемии
Талассемии – группа аутосомно-рецессивных заболеваний крови, характеризующиеся снижением выработки одного из двух разновидностей полипептидных цепей глобина (α или β), что сопровождается уменьшением наполнения эритроцитов гемоглобином и анемией.
Содержание
- Этиология
- Патогенез
- Клиническая картина
- Диагностика
- Лечение
Этиология
Талассемии подразделяют на α- и β-талассемии в зависимости от того, синтез какого пептида нарушается. Частота встречаемости β-талассемии выше, чем α-талассемий. Редкой формой является βα-талассемия.
Талассемии наиболее распространены в странах Средиземноморья, Среднего Востока и Юго-Восточной Азии.
Синтез α-цепей глобина кодируется диплоидным набором генов α α/α α (4 гена, по два от обоих родителей). Клиническая картина α-талассемии определяется числом подвергшихся делеции или мутации генов.
- Бессимптомное носительство – отсутствие клинических симптомов и изменений в гемограмме – имеет место при делеции одного из четырёх генов (- α/α α).
- Малая талассемия – незначительная анемия без признаков разрушения эритроцитов или её отсутствие, снижение MCV, MCH – формируется как результата делеции двух генов или неделетирующей мутации второго гена (- -/α α, — α/- α, — α/α α).
- Гемоглобинопатия H (промежуточная талассемия) – хроническая гемолитическая талассемия – имеет развитие при делеции трёх генов (- -/- α).
- Внутриутробная водянка – смерть во внутриутробном периоде или сразу после рождения – обусловлена делецией всех четырёх генов (- -/- -).
Выработка β-цепей шифруется двумя генами β/β (по одному от обоих родителей). В отличие от α-талассемии, при β-талассемии выработка β-цепей глобина нарушается в результате мутационного изменения функциональной активности гена. При этом возможно снижение выработки β-цепей (β+) или полное отсутствие выработки (β0).
- В соответствии с генными нарушениями выделяют несколько разновидностей β-талассемии.
- Большая талассемия (анемия Кули) имеет генотип β0/β0 с характерной клинической картиной заболевания, проявляющейся с 6–8-й недели жизни.
- При промежуточной талассемии (β+/β+ или β0/β) клиническая картина гемолитической анемии менее выражена, нуждаемость в переливаниях крови возникает при интеркуррентных инфекциях.
- Малая талассемия (β+/β) протекает без каких либо клинических проявлений, за исключением более высокой частоты анемии в период беременности.
Патогенез
Формирование проявлений заболевания и их осложнений имеют сходство среди всех типов талассемий.
Уменьшение выработки цепей глобина приводит к развитию микроцитарной анемии. Избыточные свободные цепи глобина, которые продолжают вырабатываться неповреждёнными генами, подвергаются агрегации. Образовавшиеся агрегаты неспаренных глобиновых цепей фиксируются на мембране эритроцита и повреждают её, являясь причиной преждевременной гибели эритроцита (патологический гемолиз).
В тяжёлых случаях большинство эритроидных предшественников разрушается в костном мозге (неэффективный эритропоэз). Те эритроциты, что выходят в циркуляцию, преждевременно разрушаются макрофагами селезёнки или печени.
В противовес этому срабатывают компенсаторные механизмы активации эритропоэза. Во-первых, происходит эритроидная гиперплазия костного мозга, что становится причиной расширения костномозговых полостей и деформаций, прежде всего – плоских костей скелета. Во-вторых, образуются экстрамедуллярные очаги гемопоэза в печени и селезёнке, паравертебральных зонах.
Спектр патологических нарушений при талассемиях очень широк – от едва уловимых морфологических изменений эритроцитов до состояний, опасных для жизни. Степень клинических проявлений зависит от того, какое количество генов, кодирующих цепи глобина, подвержено делеции или мутации.
Клиническая картина
Универсальными синдромами для всех клинически выраженных типов талассемий являются:
- Анемия хронического течения, вызывающая замедление роста, полового развития, хроническую сердечную недостаточность и другие, связанные с ней, осложнения.
- Гиперплазия костномозгового кроветворения, прежде всего – эритропоэза, в пределах костного скелета с возникновением очагов экстрамедуллярного кроветворения в селезёнке, печени и мягких тканях:
- расширение губчатого вещества костей черепа с рентгенологической картиной, называемой «стриженный под ёжик»;
- гипертрофия лобной кости с уплощением переносицы, сужением глазных щелей, верхней челюсти с выступанием зубов и формированием «лица бурундука»;
- расширение мозгового слоя и истончение кортикальной пластинки позвонков и длинных трубчатых костей, предрасполагающее к переломам;
- спленомегалия, гепатомегалия;
- очаги миелоидного кроветворения в мягких тканях, паравертебральных областях с возможностью сдавления спинного мозга.
- Синдром перегрузки железом из-за повышенной абсорбции железа из желудочно-кишечного тракта вследствие трансфузий эритроцитарной массы. Гемосидероз миокарда приводит к кардиомиопатии, аритмиям; гемосидероз печени – к развитию гепатоцирроза, гепатоцеллюлярной карциномы.
- Хронический гемолиз: сплено-, гепатомегалия, билирубиновые камни в жёлчном пузыре, гиперспленизм, повышающий трансфузионную зависимость.
Диагностика
- В клиническом анализе крови определяется анемия различной степени выраженности, сопровождающаяся микроцитозом и гипохромией эритроцитов, наличием мишеневидных эритроцитов и нормобластов.
- Выявленное разрушение эритроцитов сочетается с акселерированным эритропоэзом.
- Характерные изменения фракций гемоглобина, соответствующих виду талассемии.
- Типичные соматические изменения: аномалии костных структур скелета, увеличение размеров печени и селезёнки, задержка роста, полигландулярная недостаточность.
Лечение
- Тактика лечения определяется степенью выраженности клинических симптомов заболевания.
- Тяжёлые формы заболевания корригируются регулярными переливаниями эритроцитарной массы; чтобы снизить перегрузку железом при переливаниях, применяют Десферал.
- В случаях развития недостаточности эндокринных желез осуществляют заместительную гормональную терапию.
- Удаление селезёнки проводится при значительном её увеличении из-за переливаний эритроцитарной массы.
- Также необходима аллогенная пересадка костного мозга.