Агенезия мозолистого тела (АМТ) — редкое расстройство, которое присутствует при рождении (врожденное). Он характеризуется частичным или полным отсутствием (агенезией) мозолистого тела.
Мозолистое тело состоит из поперечных волокон. Причина агенезии мозолистого тела обычно неизвестна, но она может быть наследственной либо по аутосомно-рецессивному типу, либо по Х-сцепленному доминантному типу.
Кроме того, агенезия также может быть вызвана инфекцией или травмой в течение от 12 до 22 недели беременности (внутриутробной жизни), что приводит к нарушению развития мозга плода.
Внутриутробное воздействие алкоголя также может привести к АМТ.
В некоторых случаях при АМТ может возникнуть умственная отсталость, но интеллект слабо нарушенным, а так же могут присутствовать тонкие психосоциальные симптомы.
Частичная агенезия мозолистого тела
Рис.1
Полная агенезия мозолистого тела
Рис.2
АМТ часто диагностируется в течение первых 2х лет жизни. Приступы эпилепсии могут быть первым симптомом, указывающим на то, что ребенка следует тестировать на дисфункцию мозга. Расстройство также может быть без видимых симптомов в самых мягких случаях в течение многих лет.
Морфология
Классические нейрорадиологические признаки агенезии мозолистого тела:
- Передние рога и тела боковых желудочков широко расставлены и параллельны (не изогнуты). Передние рога узкие, остроугольные. Задние рога часто диспропорционально увеличены (кольпоцефалия). Вогнутые медиальные границы боковых желудочков обусловлены протрузией продольных пучков.
- III желудочек обычно дилатирован и приподнят с различной степенью дорсального расширения и смешения между боковыми желудочками. Межжелудочковые отверстия часто удлинены.
- Межполушарная борозда кажется продолжением переднего отдела III желудочка, так как отсутствует колено. В корональной проекции межполушарная борозда расширяется книзу между боковыми желудочками по направлению к крыше III желудочка. В сагиттальной плоскости обычная поясная извилина отсутствует, и средние борозды имеют радиальную или спицеобразную конфигурацию. Вокруг III желудочка часто видны межполушарные кисты. При увеличении размеров эти кисты могут приобретать аномальную конфигурацию и скрывать нижележащие пороки.
- Отсутствие мозолистого тела и прозрачной перегородки.
- Ангуляция передних рогов боковых желудочков и вдавленность их по медиальной поверхности пучками Пробста.
- Радиальный паттерн борозд и извилин отходящих от крыши III желудочка.
Признаки и симптомы
Агенезия мозолистого тела (АМТ) может первоначально проявиться началом эпилептических припадков в первые недели жизни или в течение первых двух лет. Однако не все люди с АМТ имеют судороги.
Другие симптомы, которые могут начаться в самом начале жизни, — это проблемы с кормлением и в удержании головы. Сидение, стояние и ходьба также могут быть нарушены.
Нарушение психического и физического развития и/или гидроцефалия также являются симптомами раннего начала этого расстройства.
Непрогрессирующая умственная отсталость, нарушение координации рук и глаз, а также зрительная или слуховая память могут быть диагностированы с помощью неврологического тестирования пациентов с АМТ. В некоторых легких случаях симптомы могут не появляться в течение многих лет. В легких случаях АМТ может быть упущена из-за отсутствия явных симптомов в детстве.
У некоторых пациентов могут быть глубоко посаженные глаза и выбухающий лоб. Может быть аномально маленькая головка (микроцефалия) или необычно большая головка (макроцефалия).
Складки кожи перед ушами, один или несколько согнутых пальцев (камптодактилия) и замедленный рост также могут быть связаны с некоторыми случаями агенезии мозолистого тела.
В других случаях могут присутствовать широко раздвинутые глаза, маленький нос с перевернутыми ноздрями, аномально сформированные уши, чрезмерно длинная шея, короткие руки, уменьшенный мышечный тонус (мышечная гипотония), аномалии гортани, дефекты сердца и симптомы синдрома Пьера-Робина.
Синдром Айкарди, считается наследуемым по X-сцепленному доминантному типу, состоит из агенезии мозолистого тела, инфантильных спазмов и аномалии структуры глаз.
Этот синдром является чрезвычайно редким врожденным расстройством, при котором поражаются средний слой глаз (сосудистая оболочка) и сетчатка, а также отсутствие мозолистого тела, сопровождающегося серьезной умственной отсталостью. Этим синдромом страдают только женщины.
Синдром Андермана — генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела, умственной отсталости и прогрессирующих сенсомоторных нарушений нервной системы (нейропатии).
Все известные случаи этого расстройства происходят из округа Шарлево и района Сагеней-Лак Сен-Жан в Квебеке, Канада.
Недавно был идентифицирован ген, вызывающий эту редкую форму агенезии мозолистого тела, и в настоящее время тестируется этот ген (SLC12A6).
XLAG (X-сцепленная лиссенцефалия с истинным гермофродитизмом) — является редким генетическим заболеванием, в котором самцы имеют малый и гладкий мозг (лиссэнцефалия), небольшой пенис, тяжелую умственную отсталость и резистентную эпилепсию.
Это вызвано мутациями в гене ARX. У женщин эти же мутации могут вызвать агенезию мозолистого тела самостоятельно, тогда как менее серьезные мутации у мужчин могут вызвать умственную отсталость. Диагностика это расстройства доступна в клиническом применении.
Эпидемиология
Агенезия мозолистого тела вызывает симптомы в течение первых 2х лет жизни примерно у 90% пострадавших.
Считалось, что это очень редкое состояние, но повышенное использование методов нейровизуализации, таких как МРТ, приводит к увеличению диагностики этого поражения.
Это состояние также может быть идентифицировано во время беременности с помощью ультразвука. В настоящее время самая высокая оценка заболеваемости составляет 7 на 1000 человек.
Сопутствующие нарушения
Агенезия мозолистого тела может сочетаться с расщеплением позвоночника (Spina bifida). Часть содержимого позвоночника может выступать через аномальное отверстие, что приводит к формированию менингоцеле или менингомиелоцеле.
- гидроцефалия 30%
- арахноидальные кисты
- внутричерепная липома 10%
- Мальформация Денди-Уолкера 11%
- Межполушарная киста
- Мальформация Арнольда-Киари 7%
- Срединное энцефалоцеле
- Порэнцефалия
- Голопрозэнцефалия
- Полимикрогирия
- Гетеротопия
Диагностика
Ультразвуковая (УЗИ) и магнитно-резонансная томография (МРТ).
Лечение
Лечение является симптоматическим и поддерживающим в зависимости от тяжести симптомов. Если присутствует гидроцефалия, её лечат хирургическим шунтированием снижая повышенное ВЧД. Генетическое консультирование также может принести пользу семьям с этим расстройством.
врач-рентгенолог, к.м.н. Власов Евгений Александрович
Полная или частичная перепечатка данной статьи, разрешается при установке активной гиперссылки на первоисточник
Если у вас остаются сомнения в выводах по результатам вашего МРТ — вы можете заказать пересмотр вашего исследования с подробной расшифровкой здесь:
Возможно вас так же заинтересует
Нейроэпителиальные кисты также называемые нейроглиальными или глио-эпендимными кистами, являются аномалиями развития, возникающими в результате поглощения части развивающейся нейроэктодермы, из лептоменингиальной нейроглиальной гетеротопии.
Расширенные пространства Вирхова-Робина появляются во всех возрастных группах. С возрастом пространства ВР обнаруживаются с большей частотой и большими кажущимися размерами.
Аномалия Киари — врожденное смещение структур задней черепной ямки в каудальном направлении.Основную роль сыграл Chiari, который в 1891 г. описал аномалии заднего мозга и дал их классификацию.
Цефалоцеле характеризуются дефектом свода или основания черепа, через которые выпячиваются структуры мозга. В зависимости от грыжевого черепного отверстия выделают менингоцеле, менингоэнцефалоцеле, атретическое целе и глиоцеле.
Внутричерепная липома представляет собой доброкачественное образование, которое состоит из жировой ткани. Данная патология преимущественно развивается бессимптомно. Липомы (жировики), не имеют склонности к раковой трансформации. Внутричерепная липома всегда врожденное образование и не связано с неоплазией, а является нарушением эмбриогенеза с формированием патологической области отложения жировой ткани.
Комплекс Денди-Уокера это полная или частичная агенезия червя мозжечка, вращение мозжечка против часовой стрелки с расширением IV желудочка на фоне расширения задней черепной ямки за счет смещения намета вверх.
Ромбэнцефалосинапсис — аномалия развития ромбовидного мозга (rhombencephalon). В состав ромбовидного мозга входит: мозжечок, мост и продолговатый мозг, окружающие ромбовидную ямку, которая является дном 4-го желудочка.
Голопрозэнцефалия — порок развития с нарушением формирования переднего мозга (prosencephalon), на ранних стадиях развития плода, на 3й неделе гестации (когда существуют 3-5 мозговых пузырей) при котором передний мозговой пузырь не разделен частично или полностью на два симметричных полушария.
Септооптическая дисплазия (синдром de Morsier) является одной из форм лобарной голопрозэнцефалий. Характеризуется отсутствием прозрачной перегородки, гипоплазией зрительных нервов, хиазмы и воронки.
Шизэнцефалия — расщепление коры головного мозга линейной формы или широким (но не обширным) проходом, которое распространяется от желудочков к субарахноидальному пространству.
Полимикрогирия — аномалия развития, при которой нейроны достигают коры, но вследствие неправильного распределения формируют множество мелких извилин. Наиболее часто ПМГ локализуется в области латеральной борозды (сильвиевой щели). Гистологические изменения коры могут быть выражены в различной степени.
Пахигирия — врожденный порок мозга, приводящий к сглаженности коры головного мозга . Как правило, дети с данным пороком имеют задержку развития и страдают от эпилепсии , начало и тяжесть проявления зависит от степени поражения коры головного мозга.
Изменение величины мозга — врожденная асимметрия размеров мозговых структур. Микроцефалия, или микроэнцефалия — аномально маленький головной мозг. Обычно это является результатом внутриутробного сосудистого инсульта или внутриутробной TORCH-инфекции, также причиной могут быть метаболические нарушения, в частности фенилкетонурия.
Порэнцефалия (porencephalia, греческий: poros — проход, отверстие, пора + enliephalos — головной мозг) — патологическое кистозное расширение полости желудочка головного мозга разной величины, сообщающееся с субарахноидальным пространством. В действительности граница между порэнцефалией и шизэнцефалией смазана.
Нейрофиброматоз I типа (болезнь Реклингхаузена) — наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Впервые описано во второй половине XIX века в 1882 году учеником Рудольфа Вирхова — Фридрихом фон Реклингхаузеном.
- Нейрофиброматоз II типа — наследственное заболевание с аутосомно-доминантным типом передачи, которое наследуется или возникает спонтанно, характеризующееся образованием множественных доброкачественных опухолей, преимущественно шванном и менингиом, локализующихся в центральной нервной системе и по ходу периферических нервов.
- Энцефало-тригеминальный ангиоматоз (болезнь Стерджа-Вебера, синдром Штурге-Вебера или синдром Стерджа-Вебера-Краббе) — спорадически возникающее заболевание, с возникновением ангиом мягкой мозговой оболочки и кожи лица, как правило в области глазной и верхнечелюстной ветвей тройничного нерва.
- Туберозный склероз (болезнь Бурневилля-Прингла) — генетически детерминированное заболевание, характеризующееся выраженным ростом глии в ткани мозга и сетчатки, а так же гиперплазией производных экто- и мезодермы, поражением кожи, нервной системы и наличием доброкачественных опухолей (гамартом) в различных органах.
Болезнь Гиппеля—Линдау (цереброретинальный ангиоматоз) — факоматоз, при котором гемангиобластомы мозжечка сочетаются с ангиомами спинного мозга, множественными врождёнными кистами поджелудочной железы и почек. У четверти больных развивается карцинома почки, часто первично-множественная.
Гамартома гипоталамуса — это редкое неопухолевое образование гипоталамуса. По морфологической классификации соответствует ганглиоцитоме (доброкачественная опухоль из элементов симпатических нервных ганглиев). На сегодня считается, что данное образование не представляет сложности в диагностики, но затрудняет лечение.
Общая схема форм черепа. Мезоцефалический череп (Мезокран) — нормальная анатомическая форма черепа, средняя статистически встречаемая форма — стандартная форма черепа. Долихоцефалический череп (Долихоцефал) — значительное преобладание передне-заднего размера черепа над фронтальным размером.
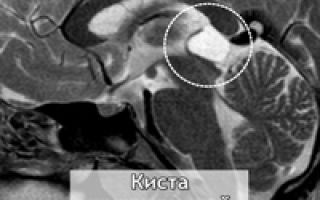
Киста шишковидной железы — доброкачественный вариант нормальной шишковидной железы. Киста шишковидной железы отличается от опухоли равномерной тонкой стенкой (до 0,2см) в которой могут быть петрификаты. Содержимое кисты может быть изоинтенсивна ликвору или серому веществу, а так же может быть гиперинтенсивна по Flair (что связано с отсутствием движения ликвора в кисте, а так же увеличенным количеством белка или дериватами гемоглобина).
Дисгенезия мозолистого тела: особенности заболевания, способы лечения и прогноз
Мозолистое тело представляет собой крепкую спайку огромных полушарий, локализованных в головном мозге. Редкой, но сложной патологией считается гипоплазия или дисгенезия мозолистого тела, которая воздействует на работу данного органа. Причины развития такой патологии могут быть различными, а эффективность проводимой терапии зависит от сложности патологии и появившихся симптомов.
Причины развития патологии
Мозолистое тело – это сплетение нервных волокон в головном мозге, соединяющее левое и правое полушария
Мозолистое тело либо большая комиссура является своеобразным скоплением нервных волокон, локализующихся в головном мозге. Они соединяют оба полушария, из которых и состоит орган.
Кроме этого, мозолистое тело отвечает за их скоординированную работу мозга, а также поддерживает процессы передачи и получения импульсов от различных полушарий. Большая комиссура связывает серое вещество, находящееся в полушариях головного мозга.
Мозолистое тело является довольно плотной по консистенции структурой, которая окрашена в белый цвет. Оно имеет вид вытянутой структуры, длина которой определяется полом пациента и его возрастом. Областью локализации большой комиссуры служит продольная щель человеческого мозга.
Зоной локализации мозолистого тела служит область, находящаяся между двумя полушариями мозга. Оно служит своеобразным проводником нейронов, и за счет его в организме человека поддерживаются следующие жизненно важные процессы:
- выражение чувств
- активные движения
- процессы познавательной деятельности
Не совсем верным служит утверждение, что при дисплазии мозолистого тела возникает резкое ограничение таких процессов. Такое состояния наблюдается лишь при осложненной форме заболевания, и наличие у пациента таких отклонений можно будет заметить невооруженным глазом.
Причина развития гипоплазии мозолистого тела еще до конца не изучена.
В то же время, большинство специалистов утверждают, что она заключается в генетических сбоях. Опасным периодом, когда имеют место такие отклонения во время внутриутробного развития плода, является первый месяц беременности.
Кроме этого, существует мнение, что причиной гипоплазии могут становиться мутации, которые воздействуют на процесс формирования мозга. В группу риска специалисты относят женщин, которые:
- распивали спиртные напитки при беременности
- подверглись радиационному облучению
- перенесли в период беременности такие патологии, как краснуха, токсоплазмоз или осложненные формы гриппа
- подверглись воздействию токсинов на организм
Дисгенезия мозолистого тела считается не распространенной и довольно сложной патологией, которая диагностируется у каждого 10-тысячного новорожденного.
Признаки и опасность забоелвания
Патология сопровождается нарушением состава и функций мозолистого тела
- Гипоплазия мозолистого тела у грудничков определяется преимущественно в течение первых нескольких месяцев после рождения, но в большинстве случаях это удается сделать во время внутриутробного развития плода.
- В ситуации, когда до появления ребенка на свет специалисты не сумели выявить патологию, в течение первых нескольких лет младенец не будет иметь каких-либо видимых отклонений в развитии.
- Лишь спустя несколько лет родители смогут заметить у их малыша следующие отклонения в развитии:
- проблемы с органами зрения
- отклонения в обонянии и осязании
- слабый крик
- инфантильность спазм
- припадки эпилепсии
- проблемы с коммуникативным общением
- судорожный синдром
- проявления, спровоцированные мышечной гипотонией
В том случае, если по каким-либо причинам не удалось диагностировать гипоплазию мозолистого тела в детском возрасте, то она обязательно проявится у взрослых людей.
Обычно возникают такие симптомы, как гипотермия, нарушение координации движений и проблемы со зрительной и слуховой памятью.
Кроме этого, часто диагностируется нарушение интеллекта, проблемы с физическим развитием и различные неврологические расстройства.
Больше и информации о строении и функциях головного мозга можно узнать из видео:
Читайте: Какая должна быть суточная норма фолиевой кислоты для беременных?
Дисплазия мозолистого тела мозга является болезнью, которая чаще всего диагностируется в период внутриутробного развития плода. В том случае, если этого не удается сделать, то при отсутствии эффективной медицинской помощи в дальнейшем наблюдается появление различных проблем в области неврологии.
Медицинская практика показывает, что у большинства пациентов с такой патологией отмечается среднее и тяжелое нарушение интеллектуального развития в сочетании с задержкой умственного и физического развития.
Кроме этого, у большей половины пациентов с гипоплазией мозолистого тела мозга выявляется такое последствие болезни, как умственная отсталость.
При не оказании эффективной помощи больному с таким диагнозом возможно развитие более серьезных нарушений психического плана и шизофрении.
Методы диагностики
Чаще всего патологию выявляют при беременности на УЗИ
Вместе с гипоплазией мозолистого тела у ребенка может выявляться большое количество различных патологий, поэтому не исключено появление дополнительной симптоматики. В большинстве случаях такую патологию удается определить в период внутриутробного развития плода с помощью УЗИ.
Для того, чтобы получить развернутую клиническую картину либо при посещении родителями специалиста могут назначаться дополнительные исследования. Проводится опрос, изучается анамнез пациента и уточняется наличие симптомов, которые свойственны для такой патологии.
Для постановки правильного и точного диагноза может назначаться:
На основе результатов полученных исследований специалист делает вывод о наличии либо отсутствии патологии, и при необходимости подбирает необходимое лечение.
Способы терапии
На сегодняшний день не существует эффективного лечения, с помощью которого удалось бы вылечить заболевание. Врачи преимущественно борются с симптоматикой дисплазии мозолистого тела, и пациенты с таким диагнозом нуждаются в постоянной терапии и поддерживающем лечении.
План лечения подбирает врач индивидуально в зависимости от тяжести поражения мозолистого тела и симптоматики заболевания. Медицинская практика показывает, что часто наблюдается неблагоприятный исход. У детей с таким диагнозом высока вероятность развития различных психических нарушений и шизофрении.
Способы и методы лечения зависят от тяжести патологии
Лечение патологии проводится с помощью терапевтического вмешательства, которое можно условно разделить на несколько групп:
- симптоматическое воздействие на пациента
- назначение заместительных и консервативных вариантов лечения хронической недостаточности почек
В том случае, если выявляется аномалия структурного и анатомического характера, то прибегают к проведению хирургического вмешательства. Медицинская практика показывает, что если правильно и профессионально провести операцию, то эффект терапии будет положительным.
Чаще всего к проведению операции прибегают в том случае, когда появляются опасные осложнения в жизненно важных органах человека, имеющих непосредственное отношение к мозолистому телу. Важную роль играет и тот факт, насколько вовремя было проведено хирургическое вмешательство.
Советы родителям
При постановке такого диагноза, как гипоплазия головного мозга, малышу понадобиться помощь и поддержка родителей. В домашних условиях можно воспользоваться следующими рекомендациями:
- контролировать состояние ребенка и в том случае, если он устал, позволить ему немного передохнуть, после чего продолжить занятие
- носить малыша в позе самолета, которая оказывает укрепляющее действие на детский организм
- выкладывать ребенка лицом к лицу себе на грудь и гладить его от головы к попке, что позволит перенести вес от головы к тазу
- при произнесении ребенком звуков следует обязательно их повторять с такой же интонацией, выдерживая небольшие паузы
- при играх с погремушкой нужно дать ребенку возможность зафиксировать на ней взгляд, не спеша водить ее из стороны в сторону
Гипоплазия мозолистого тела является серьезной патологией, которая часто сопровождается развитием сопутствующих патологий. Родителям необходимо набраться терпения и обязательно настроиться на благоприятный исход.
Агенезия мозолистого тела :: Симптомы, причины, лечение и шифр по МКБ-10
Агенезия мозолистого тела
Агенезия мозолистого тела. Это врожденное отсутствие мозолистого тела или его части. Аномалия вызвана генетическими нарушениями, пороками развития сосудов, тератогенными факторами. Основные признаки заболевания: двигательные нарушения, задержка умственного развития, судороги. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначают КТ или МРТ головного мозга, нейросонографию у новорожденных, генетические исследования. Симптоматическое лечение: медикаментозная коррекция осложнений, реабилитационные программы.
Агенезия мозолистого тела
Агенезия мозолистого тела (ТМА) — одно из наиболее частых пороков развития нервной системы. Распространенность заболевания в популяции составляет от 0,05% до 7% у новорожденных, а в группе детей с задержкой развития психики агенезия встречается у 2,3%.
Калифорнийская программа по врожденным дефектам предусматривает другой уровень агенезии — 1,4 на 10 000 живорождений. Впервые это состояние было описано в 1812 году на вскрытии, проведенном немецким анатомом И.
Рэйлем, и названо «естественной моделью рассеченного мозга».
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает многофакторная теория, согласно которой для формирования врожденной аномалии ЦНС необходимо сочетание неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенеза: • Генетические аномалии.
Поражение мозолистого тела отмечается при различных наследственных синдромах: Миллера-Диккера, Рубинштейна-Тауби, Доннея-Кугана. Заболевание является частью как минимум 7 аутосомно-доминантных заболеваний, 23 аутосомно-рецессивных и 12 X-сцепленных врожденных заболеваний. • Сосудистые расстройства.
Причиной недоразвития мозолистого тела могут быть артериовенозные мальформации или аневризмы, для которых характерно отсутствие нормальной капиллярной сети. В этом случае происходит явление кражи, МТ-клетки не получают должного количества кислорода и питательных веществ. • Токсические эффекты.
Заболевание связано с действием тератогенных химических факторов: лекарств, солей тяжелых металлов, пестицидов и бытовой химии. Вдыхание табачного дыма (активное или пассивное курение) или прием алкоголя беременной женщиной во время вынашивания отрицательно влияет на формирование центральной нервной системы плода. • Внутриутробные инфекции.
Аномалии формирования неврологических структур, в том числе агенезия мозолистого тела, возникают при попадании возбудителей в плод на 2–3 мес беременности. Герпетические инфекции, токсоплазмоз, цитомегаловирус проявляют нейротропные свойства. Главный фактор риска — недоношенность.
У новорожденных, родившихся до 27 недель беременности, МТ истончается в задних отделах, между 28 и 30 неделями — только в области гребня. У рожденных после 30 недель в неонатальном периоде изменений не обнаруживается, хотя нейропсихологическое исследование у школьников часто выявляет дефицит межполушарной передачи когнитивной информации.
Мозолистое тело (МТ) представляет собой большой пучок комиссуральных нервных волокон. Это важный аксональный путь, соединяющий соответствующие области коры правого и левого полушарий. Анатомическая структура длиной 7-9 см состоит из более чем 300 миллионов аксонов. Формирование МТ начинается на стадии позднего нейроонтогенеза (8-9 недель эмбриогенеза). Созревание обычно длится до 20-25 лет. Агенезия наступает при нарушении дифференцировки нервной трубки в период от 2 до 5 месяцев внутриутробного развития. При полном отсутствии МТ третий желудочек мозга остается открытым, столбы свода не сформированы, прозрачные перегородки отсутствуют. В 60% случаев при АМТ передняя спайка отсутствует. В 10% он увеличен и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах послеродового периода.
Характерным анатомическим изменением является кольпоцефалия, при которой расширяются задние отделы боковых желудочков головного мозга.
Состояние не относится к истинной гидроцефалии у новорожденных, но связано с уменьшением ассоциативных путей коры головного мозга.
Другой типичный признак дефекта — пучки Пробста, которые представляют собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
В практической неврологии состояние делится на полное, когда орган полностью отсутствует, и частичное (частичное), при котором методы визуализации не обнаруживают отдельные участки ТМ. Это важно из-за тяжести клинической картины и возможных осложнений.
В соответствии с патогенетическими особенностями формирования врожденных пороков развития выделяют следующие 3 формы заболевания: • Агенезия. Очертание зародышевого зачатка МТ полностью отсутствует. • Аплазия. Зачаток мозолистого тела присутствует, но не развивается. • Гипоплазия.
ТМ недоразвита из-за нарушений на одном из этапов эмбриогенеза: уменьшаются размеры и масса органа, снижается его функциональная активность.
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (с гипоплазией) до критических психоневрологических расстройств с грубым недоразвитием, сопровождающимся другими врожденными пороками центральной нервной системы. У новорожденных признаки патологии могут полностью отсутствовать и появляться по мере взросления малыша с задержкой психомоторного развития. Двигательные расстройства определяются у 35-40% пациентов. Они проявляются гипотонией или дистонией мышц, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Позже дети начинают держать голову, им трудно научиться сидеть, ползать и ходить. Могут отмечаться нарушения координации, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги. Мозолистое тело поддерживает связь между областями мозга, образует межполушарную организацию высших психических процессов. При его агенезии или гипоплазии у детей выявляются когнитивные нарушения. У новорожденных и в раннем детстве наблюдается задержка речи, снижение динамической составляющей игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, нарушение памяти, при общей АМТ коэффициент интеллекта снижается.
Ассоциированные симптомы: Нарушение понимания речи. Нарушение походки. Отсутствие речи. Судороги. Судороги в ногах.
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают пороки развития коры головного мозга (22,8%), межполушарные кисты (14,3%), голопроэнцефалия (14,3%). Менее распространенными сопутствующими аномалиями являются кисты и гипоплазия мозжечка, синдром Арнольда-Киари.
Помимо структур центральной нервной системы, до 20% новорожденных имеют дефекты ряда внутренних органов. Симптоматическая эпилепсия височно-лобной локализации наблюдается у 75% пациентов с тотальным поражением, а когнитивные нарушения возникают в 66% случаев. У 16% пациентов развиваются расстройства аутистического спектра.
Изредка возникают патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанных аномалий зрительных нервов.
Акушерское УЗИ — основной метод обследования в дородовой период. У новорожденных нейросонография используется для скринингового диагноза, но этот метод не всегда показывает хорошую информативность, особенно в случаях частичной агенезии. Для проверки диагноза назначаются следующие методы исследования: • КТ головного мозга.
С помощью компьютерной томографии определяют широко расставленные передние рога, приподнятое положение третьего желудочка, параллельный ход медиальных стенок боковых желудочков. КТ выполняется в рамках послеродовой диагностики. • МРТ головного мозга.
Для наиболее точной визуализации степени агенезии или гипоплазии мозолистого тела новорожденному проводят трехплоскостную магнитно-резонансную томографию. В зависимости от показаний беременным может быть рекомендована МРТ для исключения сопутствующих нарушений ЦНС, несовместимых с жизнью. • Нейропсихологическое обследование.
Для изучения когнитивных функций у детей используются Шкала интеллекта Векслера (пересмотренная WISC), оцениваемые тесты по чтению и правописанию Шеннеля, оценка беглости речи и тест на ассоциативную связь с контролируемым устным словом. • Генетический анализ.
Для подтверждения или исключения наследственных заболеваний, сопровождающихся агенезом мозолистого тела, представлены результаты кариотипа и секвенирования генома как у новорожденных, так и у детей разного возраста. Также проводятся исследования в пренатальный период, чтобы решить, сохранить ли беременность или прервать ее.
Специфической терапии нет. Медикаментозное лечение назначает индивидуально неонатолог или педиатр с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста применяются противосудорожные препараты, нейрометаболические препараты и дегидратационная терапия.
В основе медицинской помощи лежит комплексная реабилитация, которая состоит из следующих элементов: • Неврологические программы. Занятия с детским логопедом направлены на развитие речевых функций, устранение симптомов дизартрии и улучшение артикуляции.
• Дефектологические программы. В случае детей с ограниченными интеллектуальными возможностями, которых нельзя обучать в обычных классах, требуется помощь педагогов. • Нейроакустические программы.
Формирование и гармонизация высших психических функций происходит с помощью звуковой и музыкальной терапии.
Прогноз зависит от вида врожденной аномалии мозолистого тела, наличия сопутствующих пороков развития центральной нервной системы.
Благоприятный исход наблюдается при частичной гипоплазии МТ, а при сложных пороках развития головного мозга у новорожденных могут быть опасные для жизни осложнения.
Профилактические меры включают медико-генетическое консультирование, исключение тератогенных эффектов при беременности.
1. Эпилептические проявления когнитивные и аутистические расстройства у пациентов с агенезией мозолистого тела: результаты нейропсихологического тестирования/ О.А. Милованова, О.А. Комиссарова, Т.Ю. Тараканова, С.В. Бугрий// Эпилепсия и пароксизмальные состояния. — 2018. — №4. 2. Влияние особенностей строения мозолистого тела и доминирующего полушария на протекание психических процессов в юношеском возрасте/ У.С. Чернышова, Т.Ю. Хабарова, Д.А. Соколов// Центральный научный вестник. — 2016. 3. Нейропсихологический анализ патологии мозолистого тела/ М.С. Ковязина. — 2016.
4. Молекулярная эмбриология: на пути каталогизации генов врожденных пороков развития головного мозга/ В.П. Пишак, М.А. Ризничук// Международный журнал педиатрии, акушерства и гинекологии. — 2014. — №5.
42a96bb5c8a2acfb07fc866444b97bf1 Модератор контента: Васин А.С.
Детский аутизм, ассоциированный с неврологическими проявлениями и гипоплазией мозолистого тела
Детский аутизм относится к нарушениям психического развития, протекающим с расстройством социальных и коммуникативных функций, снижением физической активности, со склонностью к двигательным и/или речевым стереотипиям.
Авторы статьи провели исследование с целью обобщения данных о клинико-диагностических особенностях и демонстрации редких наследственных синдромов, протекающих с клинической картиной детского аутизма, неврологическими расстройствами и структурно уменьшенным в размере МТ.
Диагностика детского аутизма включает анализ анамнестических данных, медико-генетические, неврологические, психопатологические, патопсихологические, лабораторные и инструментальные обследования. Ранняя диагностика ГМТ возможна на сроке беременности 18–20 нед. При подозрении на ГМТ у плода проводят пренатальное кариотипирование.
На пренатальных диффузионно-тензорных МР-изображениях клюв и колено МТ практически сформированы к 15-й неделе гестации. Окончательное строение МТ приобретает к 20-й неделе гестации. На постнатальных МР-изображениях ГМТ визуализируется в виде равномерно истонченного контура с уменьшением его размера в переднезаднем направлении.
Определенный интерес у больных с ГМТ представляет сочетание неврологических и психических симптомов.
Особенностью аутизма в раннем возрасте является неравномерное накопление лексического словаря; отсутствие подражательной функции и недостаточная коммуникативная направленность речи.
У ряда пациентов отмечается общее недоразвитие речи; нарушения поведения; отсутствие реакции на собственное имя при анатомически неповрежденном слуховом анализаторе; гиперсенситивная реакция на свет, звуки и тактильные стимулы; могут наблюдаться эпилептические приступы, двигательные расстройства.
У пациентов старшего возраста, как правило, сохраняется интеллектуальная недостаточность вплоть до умственной отсталости; эпилептические приступы, входящие в структуру эпилепсии; слабая или практически отсутствующая дифференцированность эмоциональных проявлений; несформированность социально-коммуникативных навыков; недостаточное использование парадигматических средств общения (мимика, жесты); двигательные расстройства.
К настоящему времени установлено, что детский аутизм в сочетании с ГМТ ассоциирован с различными нейрогенетическими синдромами, характеризующимися неспецифичностью клинических проявлений, особенно на ранних стадиях болезни.
Специалисты проанализировали клинические случаи — у двух обследованных пациенток с нейрогенетическими синдромами, сопровождающимися на МРТ головного мозга гипоплазией МТ, были изучены психические и неврологические нарушения. Проведены комплексный анализ анамнестических данных; медико-генетическое, неврологическое, психопатологическое, патопсихологическое, лабораторные и инструментальные обследования.
У первой пациентки выявлен нейрогенетический синдром Вольфа-Хиршхорна, (46,XХ,t(4;6)(p16.3;p23),del(4)(p16.3)), у второй – синдром Прадера-Вилли, (46,XХ 15q11.2q13).
В представленных двух наблюдениях отмечен отягощенный акушерский анамнез, неблагополучный анте и интранатальный периоды: угроза прерывания в I триместре, задержка внутриутробного развития плода в III триместре, преждевременные оперативные роды на сроке гестации 31 нед посредством экстренной операции кесарева сечения в связи с остро возникшей внутриутробной гипоксией плода в первом наблюдении; во втором: гестоз беременных в III триместре, экстренная операция кесарева сечения на сроке 34–35 нед. (преждевременная отслойка плаценты).
Обе пациентки имели низкую массу тела при рождении, признаки гипоксически-ишемического поражения головного мозга, более выраженные у первой пациентки (оценка по шкале Апгар 5/6 баллов, морфофункциональная незрелость, недоношенность 2-й степени), во втором случае – 7/8 баллов, недоношенность 1-й степени.
В дальнейшем низкие показатели оценки состояния анте-, интра- и постнатального периодов коррелировали с выраженностью психических, неврологических и когнитивных расстройств.
К наиболее ярким системным клиническим проявлениям дизэмбриогенетических синдромов относятся психические и неврологические расстройства. В данной работе выраженность аутистических расстройств зависела от степени поражения головного мозга.
В качестве иллюстрации тяжелого течения детского аутизма – первый клинический случай – больная девочка с тяжелой степенью умственной отсталости, тотальным недоразвитием всех уровней психической деятельности, отсутствием вербальных средств общения, двигательными стереотипиями, нарушением социального взаимодействия в сочетании с ранним органическим поражением головного мозга.
Второй случай – органический аутизм с умственной отсталостью легкой степени с негрубым нарушением социальной адаптации, недостаточной сформированностью отдельных структур 1-го и 2-го функционального блока мозга.
Эпилептические проявления у первой пациентки представлены в виде структурной фокальной эпилепсии, подтвержденной результатами ЭЭГ (мультирегиональная эпилептиформная активность); у второй пациентки зарегистрирована непостоянная региональная эпилептиформная активность в лобно-височных отведениях слева, на момент осмотра, не требовавшая назначения противоэпилептической терапии. Важно отметить, что у первой больной выявлена тяжелая задержка психомоторного развития, предшествующая манифестации эпилептических приступов, что говорит о формировании выраженной церебральной патологии в дебюте заболевания. В первом случае на МРТ верифицирована гипоплазия задних отделов МТ, умеренные постгипоксические изменения вещества головного мозга; во втором – гипоплазия перешейка МТ, кортикальная субатрофия лобных долей, негрубое расширение наружных субарахноидальных пространств, ограниченные зоны перивентрикулярного глиоза.
У первой пациентки выявлен когнитивный дефицит, соответствующий тяжелой степени умственной отсталости; у второй пациентки – преобладали негрубые нарушения зрительно-пространственной координации, что коррелировало с целостностью задних отделов МТ.
Становится понятным, что уровень когнитивного дефицита может быть в некоторой степени величиной, связанной с толщиной заднего отдела МТ.
Это имеет свое объяснение: более высокая степень миелинизации нервных волокон способствует большей скорости распространения нервных импульсов по нервным волокнам, тем самым обеспечивая наилучшие результаты у обследуемых пациентов.
Таким образом, тяжесть клинической картины у больных девочек обусловлена наличием дизэмбриогенетических нарушений в сочетании с перинатальными осложнениями. В настоящее время специфическая терапия нейрогенетических синдромов не разработана.
Тактика ведения подобных пациентов зависит от тяжести состояния, течения заболевания и включает в себя проведение регулярных реабилитационных мероприятий (массаж, лечебная физкультура), назначение симптоматической терапии, направленной на уменьшение выраженности проявлений спастичности.
Важным компонентом лечения подобных больных является медико-психолого-педагогическое сопровождение, направленное на уменьшение выраженности аутистических расстройств.
Длительное воздействие неблагоприятных перинатальных факторов, способствующих нарушению нормального развития мозга, является одной из причин формирования ГМТ.
Основное внимание в диагностике нейрогенетических синдромов принадлежит комплексному неврологическому, психиатрическому обследованию, нейропсихологическому тестированию, ЭЭГ-диагностике и нейровизуализационным техникам.
Для окончательного подтверждения диагноза требуется ДНК-тестирование.
Динамика интеллектуальных расстройств при детском аутизме зависит от характера заболевания, в структуре которого наблюдается данное расстройство, одним из звеньев патогенеза которого является повреждение высших психических функций на этапах раннего онтогенеза.
Относительно благоприятный прогноз заболевания отмечается при более позднем дебюте неврологического и психического дефицита.
В структуру различных наследственных нейрогенетических синдромов входит сложная комбинация неврологических, психических, нейрофизиологических и радиологических нарушений; их своевременное распознавание требует большого клинического опыта, практических знаний не только в области неврологии, но и в смежных разделах медицины.
Источник: Детский аутизм, ассоциированный с неврологическими проявлениями и гипоплазией мозолистого тела: обзор литературы и клинические случаи Комиссарова О.А., Милованова О.А., Авакян Г.Г., Бугрий С.В.
Том 12, № 1 (2020)