Фенилкетонурия представляет собой достаточно тяжелое наследственное заболевание, основная тяжесть проявлений которого сосредоточена, прежде всего, на нервной системе. Фенилкетонурия, симптомы которой чаще всего встречаются среди девочек, возникает по причине нарушения обмена аминокислот, учитывая же поражение при этом ЦНС, ее проявления сводятся к нарушению умственного развития.
Общее описание
В классической своей форме, которая актуальна для большинства случаев, фенилкетонурия, которую также принято определять как фенилпировиноградную олигофрению, связана с резкостью снижения активности печеночного фермента. Им в частности является фениланин-4-гидроксилаз.
Нормальное состояние организма предусматривает катализацию им превращения в тирозин фенилаланина. 1% случаев отмечается возникновением атипичной формы фенилкетонурии, которая возникает по причине мутаций в другого типа генах, которые ответственны за процесс кодирования ферментов.
Наследование заболевания происходит в соответствии с аутосомно-рецессивной схемой.
Что касается процессов, происходящих при рассматриваемом заболевании, то они заключаются в следующем. Возникновение характерного метаболического блока провоцирует активацию побочных путей по обмену фенилаланина, что приводит к накоплению токсичных производных от его действия.
К таким производным в частности относятся фенилмолочная и фенилпировиноградные кислоты, практически не образующиеся при нормальном состоянии организма. Они же влияют на ЦНС, провоцируя нарушения белкового обмена, обмена липопротеидов и обмена гликопротеидов.
Одновременно с этим возникают расстройства в транспорте аминокислот, нарушения в обмене серотонина и катехоламинов, актуальность приобретают также и перинатальные факторы.
Помимо этого начинают также образовываться и в норме практически отсутствующие ортофенилацетат и фенилэтиламин. Их избыток провоцирует нарушения в метаболизме липидов, происходящем в головном мозге.
Как предполагается, именно это становится причиной прогрессирующего состояния снижения у больных данным заболеванием интеллекта, что может достичь даже идиотии.
В целом же окончательно механизм, в соответствии с действием которого происходит нарушение в развитии функций мозга в случае с актуальным для организма заболеванием фенилкетонурией, не ясен.
Фенилкетонурия: симптомы заболевания
Здесь, прежде всего, важно отметить, что первые недели жизни ребенка не позволяют внешне определить данного заболевания. Первые его признаки проявляются через два-шесть месяцев с момента рождения малыша.
Он становится вялым, наблюдается отсутствие заинтересованности в отношении условий, его окружающих и мира в целом. Также ребенок становится беспокойным, нарушениям подвергается мышечный тонус. Появляется рвота, судороги, тяжелые кожные экземы.
Под экземами в частности подразумевается острая или хроническая форма воспалительного и незаразного заболевания, при котором образуется сыпь. Природа заболевания аллергическая, дополнительными проявлениями симптоматики выступает чувство жжения и выраженный зуд кожи.
Актуальна и склонность к рецидивам, то есть к повторному возникновению симптоматики после относительного временного его затишья.
Шестой месяц позволяет определить отставание в развитии у ребенка. Одновременно с этим теряется способность к фокусированию взгляда на отдельных предметах, малыш перестает узнавать родителей.
Отсутствует реакция на цветные/яркие игрушки.
Важно оперативно приступить к лечению, в противном случае отсталость развития будет постепенно подвергаться лишь прогрессированию в актуальных для нее процессах.
Физическое развитие больных младенцев на физическом уровне отмечается меньшими нарушениями, нежели на психическом. В обхвате голова может быть несколько меньших размеров, чем это предусмотрено для показателей нормы. Зубки прорезываются позже, позже ребенок начинает сидеть и ходить.
Принятие положения стоя у таких малышей сопряжено с широким расставлением для этого ног, а также со сгибанием их в коленях и в тазобедренных суставах, плечи и голова при этом опущены. Что касается особенностей ходьбы у больных детей, то она характеризуется покачиваниями, шажки небольшие.
Сидят дети, поджав под себя ножки, что обуславливается значительным мышечным тонусом, который они испытывают.
Отличаются детки и характерной внешностью со светлыми волосами, кожа у них абсолютно белая, без пигментации, глаза светлые. Учитывая излишнюю белизну кожи, она нередко покрывается у детей сыпью, что объясняется ее особой чувствительностью в отношении воздействия ультрафиолетового излучения.
В числе основных проявлений, свойственных фенилкетонурии, можно также выделить характерный «мышиный» запах, в некоторых случаях возможны эпилептические припадки, которые, однако, с возрастом исчезают.
Выраженными проявлениями выступают синюшность конечностей, дермографизм (изменение в окраске кожи местного типа, происходящее при механическом воздействии на нее), потливость.
Чаще среди больных фенилкетонурией отмечается, помимо перечисленных симптомов, наличие артериальной гипотонии, дерматита, частых запоров, тремор (т.е. дрожание), потеря равновесия и расстройство в виде нарушения координации движений.
Диагностирование фенилкетонурии
Важным, как мы уже отметили, является раннее диагностирование заболевания, что позволит избежать его развития и привести к ряду необратимых и тяжелых последствий. По этой причине в родильных домах к 4-5 дням жизни (для новорожденных доношенных) берется для анализа кровь. У недоношенных детей на предмет фенилкетонурии (ФКУ) кровь берется на 7 день.
Процедура предусматривает взятие капиллярной крови по прошествии часа с момента кормления, ею в частности пропитывается специальный бланк. Концентрация, указывающая на отметку свыше 2,2% фенилаланина в крови малыша, требует направления его с родителями для осмотра в медико-генетический центр. Там же проводится дообследование и, собственно, уточнение диагноза.
Причины фенилкетонурии
Фенилкетонурия может быть спровоцирована следующими факторами:
- Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием;
- Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.
Сам процесс наследования гена ФКУ может носить случайный характер, что в качестве примера отражено в приведенной ниже схеме.
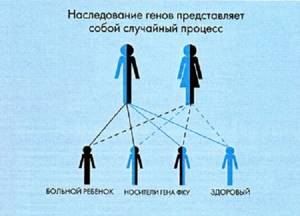
Возможность наследования генов ФКУ
Лечение фенилкетонурии
Единственный метод лечения заключается в своевременной организации диетотерапии, которая требуется с первых дней жизни. Заключается она в резком ограничении фенилаланина, который содержится в отдельных пищевых продуктах. Таким образом, исключаются все белковые продукты.
Учитывая продолжительность и полное исключение фенилаланина из пищи, возможным становится расщепление собственных белков, что ведет к истощению организма больного. По этой причине потребность в получении белка возмещается за счет аминокислотных смесей и белковых гидролизатов.
Со сменой снижения концентрации в крови фенилаланина до нормальных показателей, в рацион постепенно добавляются продукты животного происхождения. В рацион добавляются различные фрукты и сезонные овощи, растительные и животные жиры, а также углеводы.
Естественно, что контроль над содержанием фенилаланина следует продолжать вести. Организм должен быть снабжен данной аминокислотой в должном объеме для обычного развития и роста малыша, однако, за исключением возможности скапливания его в тканях.
Строжайшая диета соблюдается на протяжении, как минимум, пяти лет. Что касается возраста более взрослого, то здесь в значительной степени снижается подверженность нервной системы к воздействию, оказываемому фенилаланином, а также воздействию, оказываемому продуктами его распада. При соблюдении требуемых мер к 12-14 годам ребенок сможет свободно перейти к обычному питанию.
Примечательно, что медикаментозное лечение при данном заболевании носит синдромный характер.
В него включено применение препаратов, ориентированных на устранение судорог, в том числе и препаратов, которые оказывают стимулирующее воздействие на интеллектуальную деятельность.
В обязательном порядке детям назначается курс лечебной физкультуры и массажа. Дополнительно назначаются уроки, способствующие развитию логики.
Диагностирование фенилкетонурии производится педиатром и генетиком на основании специальных анализов в комплексе с общей характерной симптоматикой.
Источник: https://SimptoMer.ru/bolezni/nevrologiya/49-fenilketonuriya-simptomy
Фенилкетонурия
«Помню, когда ей было три месяца, она лежала в своей маленькой корзинке на прогулочной палубе корабля. Пока мы путешествовали, я приносила ее сюда, чтобы она дышала утренним воздухом. Люди, прогуливающиеся по палубе, останавливались взглянуть на нее, и меня одолевала гордость, когда они говорили о ее необычной красоте и о разуме в ее глубоких голубых глазах», — так писала о своей первой дочери Кэрол — американская писательница Перл Бак (“The Child Who Never Grew”, 1950). Автор длительно вынашивала идею написать это произведение не только для того, чтобы выразить свою боль, но и помочь другим родителям, находящимся в подобной ситуации. Но можно сказать, что эта новелла стала, вероятно, первым описанием ребенка с далеко не редкой болезнью: в 1960 году Кэрол, сильно отстающей в развитии и обучающейся в специальной школе, поставили диагноз «фенилкетонурия».
Хотя все началось несколько раньше…
В 1934 году физиолог Асбьерн Феллинг, изучавший метаболические расстройства, определил причину необычного запаха мочи у двух норвежских детей с умственной отсталостью: виной тому был избыточный уровень одного из метаболитов фенилаланина — фенилпировиноградной кислоты.
Год спустя британцем Пенроузом был предложен термин «фенилкетонурия», а также определен аутосомно-рецессивный тип передачи заболевания. Помимо этого, Пенроуз предложил лечебную диету, но она не была принята. Аналогичная идея, озвученная Джервисом и Бикелем несколько позже, уже в 50-х, стала и остается до сих пор краеугольным камнем в лечении ФКУ.
В 60-х микробиолог Роберт Гатри предложил диагностический тест для определения гиперфенилаланинемии: в качестве индикатора он использовал колонии Bacillus subtilis, которым для роста необходим фенилаланин.
В наши дни многие страны по всему миру включили тест Гатри (либо более новые тестовые системы, основанные на тандемной масс-спектрометрии) в программы неонатального скрининга, что позволило сразу же приступить к лечению новорожденных и избежать серьезных нарушений интеллекта.
Последние 20 лет прошлого века пролили свет на генетическую природу ФКУ, а в конце первого десятилетия 21-го века была сформирована база данных мутаций гена фермента фенилаланингидроксилазы, являющихся причиной развития заболевания. Примерно в это же время были установлены генетические причины нарушения метаболизма тетрагидробиоптерина.
Итак…
Фенилкетонурия (ФКУ) — врожденное нарушение метаболизма фенилаланина, приводящее к избыточному накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии, ГФА) и его дериватов.
Наиболее часто (~ 97–98 %) развитие ФКУ обусловлено мутацией гена фенилаланингидроксилазы (ФАГ), локализованного на длинном плече 12 хромосомы, участке 12q22–q24.1, которая наследуется аутосомно-рецессивно.
Данный фермент лимитирует реакцию превращения фенилаланина в тирозин, и уровень ГФА, и, соответственно, тяжесть заболевания напрямую зависят от его активности, которая определяется особенностями мутации гена.
В остальных ~ 2–3 % случаев ФКУ вызвана недостаточностью тетрагидробиоптерина, которая развивается из-за мутацией гена одного или нескольких ферментов, регулирующих его обмен (BH4-дефицитная ФКУ). BH4 является коферментом ФАГ, а также некоторых других энзимов, опосредующих синтез дофамина и серотонина (см. рис.1).
В МКБ-10 выделяют «классическую ФКУ» и «другие гиперфенилаланинемии».«Классический» вариант заболевания дифференцируется по степени тяжести согласно уровню фенилаланина в крови (см. табл.1)
Таблица 1 | Классификация классической ФКУ по степени тяжести
| Форма ФКУ* | Уровень фенилаланина в крови, мкмоль/л | Уровень фенилаланина в крови, мг/дл |
| Легкая ГФА** (не ФКУ) | 120–600 | 2–10 |
| Умеренная (мягкая, средняя) | 600–1200 | 10–20 |
| Классическая (тяжелая) | >1200 | >20 |
ФКУ* — фенилкетонурия; ГФА** — гиперфенилаланинемия
Благодаря результатам генетических исследований была создана классификация, отражающая этиопатогенез ГФА и ФКУ (см. табл.2)
Таблица 2 | Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии
| Название | Причинный фермент |
| ФАГ*-зависимая ФКУ** | Фенилаланин-4-гидроксилаза |
| ГФА***, BH4****-дефицит, тип А (ФКУ, 3 типа) | 6-пирувоил-тетрагидроптерин синтаза |
| ГФА, BH4-дефицит, тип B | Гуанозинтрифосфат-циклогидролаза |
| ГФА, BH4-дефицит, тип C (ФКУ, 2 типа) | Дигидроптеридинредуктаза |
| ГФА, BH4-дефицит, тип D | Птерин-4-альфа-карбиноламиндегидратаза |
| ГФА, BH4-дефицит | Сепиаптеринредуктаза |
ФАГ* — фенилаланингидроксилаза; ФКУ** — фенилкетонурия; ГФА*** — гиперфенилаланинемия; BH4**** — тетрагидробиоптерин
Другие ГФА встречаются как при физиологических, так и при патологических состояниях.
У новорожденных может быть транзиторное повышение уровня фенилаланина в крови до патологических значений ввиду незрелости ферментных систем печени или избыточного белкового питания матери, но, как правило, состояние это не длительно, а клинические проявление незначительны либо вовсе отсутствуют. Патологическая ГФА может сопровождать поражения печени различной этиологии и в этом случае будет имеет вторичный характер.
Патогенез
Фенилаланин является незаменимой аминокислотой, поступающей в организм человека преимущественно в составе белковых продуктов животного происхождения. Большая часть этой аминокислоты расходуется на синтез собственных белков организма, а оставшаяся часть — на синтез тирозина, что является главным путем катаболизма фенилаланина.
Эта реакция регулируется ферментом ФАГ при участии кофермента BH4 (см. рис 1). Отсутствие данного энзима либо его малое количество (при ФКУ от 0 до 50 % нормальной активности фермента) приводит к накоплению фенилаланина и развитию клинической картины ФКУ различной степени тяжести.
Не утилизированный фенилаланин катаболизируется по минорному пути с образованием токсичных продуктов (фенилацетата, фенилпирувата,фениллактата), а сниженное образование тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов (меланинов).
Помимо участия в синтезе тирозина, BH4 является коферментом в реакциях образования ДОФА и серотонина. Также на количество медиаторов ЦНС влияет и само количество фенилаланина.
Дело в том, что в норме фенилаланин, а также тирозин (как уже было обозначено выше — предшественник дофамина, норадреналина и адреналина) и триптофан (предшественник серотонина) преодолевают гематоэнцефалический барьер при помощи переносчика больших нейтральных аминокислот LAT1. Возросший при ФКУ уровень фенилаланина может ингибировать LAT1, препятствуя поступлению иных субстратов в нейроны.
Рисунок 1 | метаболизм фенилаланина
Клиническая картина
Первые симптомы нелеченной ФАГ-зависимой ФКУ появляются, как правило, на первом году жизни ребенка, достигая максимума ко второму полугодию. Сперва обращает на себя внимание вялость ребенка либо, напротив, его беспокойство, возбужденность и срыгивания, нарушение мышечного тонуса, судороги, а также специфический затхлый запах мочи, названный «мышиным».
Кроме того, нередко ФКУ проявляется эпилептическими приступами в виде абсансов, кивков, генерализованных судорог. Несколько позже, по мере роста ребенка, становится очевидным его задержка в моторном и нервно-психическом развитии.
Болезнь, при отсутствии лечения, прогрессирует медленно, но неуклонно, приводя к глубокой олигофрении, несформированности речи, отсутствию игровой и предметной деятельности. Фенотипически для детей и взрослых, больных ФКУ, характерна гипопигментации кожи, волос и радужки.
При BH4-дефицитной ФКУ, помимо вышеобозначенных признаков, из-за большей недостаточности нейротрансмиттеров ЦНС выявляются атаксия, тремор, нарушения мышечного тонуса, гипокинезия, нарушения терморегуляции, затруднение глотания и поперхивания.
Диагностика
Первый этап лабораторной диагностики проводится на 3–7-й день жизни (но не ранее, чем через 2 дня от начала энтерального питания) новорожденного в рамках неонатального скрининга путем определения уровня фенилаланина на сухом пятне крови с помощью флюориметрии или тандемной масс-спектрометрии.
При ГФА (фенилаланин > 120 мкмоль/л или > 2 мг/дл) проводится ретест. Если при повторном исследовании были получены подобные результаты, переходят ко второму этапу — определению отношения фенилаланин/тирозин.
Этот косвенный метод позволяет провести дифференциальную диагностику между ФАГ-зависимой и BH4-зависимой ФКУ, что важно для назначения правильного лечения. Кроме лабораторных методов с целью уточнения типа заболевания используют молекулярно-генетические методы.
При отсутствие возможности провести неонатальный скрининг, в постановке диагноза опираются на клиническую картину, биохимические показатели, генеалогический анамнез, молекулярно-генетическую диагностику.
- При выявлении легкой ГФА необходимо дальнейшее наблюдение и повторная диагностика.
- Лечение
- Основная цель терапии ФКУ — снижение уровня фенилаланина в крови для избежания нарушения моторного и нервно-психического развития ребенка — , достигается следующими методами:
- Гипофенилаланиновая диета — основной способ лечения уже более 60 лет. Для уменьшения поступления фенилаланина больным следует ограничивать прием высокобелковой пищи (мясо, рыба, яйца, молочные продукты, орехи, бобовые и др.) и вводить в рацион растительные продукты с высоким содержанием тирозина. Строгость диеты напрямую зависит от степени ГФА, меню должно составляться с опорой на факт «1 г белка = ~ 50 мг фенилаланина», возрастные физиологические нормы потребности в фенилаланине, тирозине и соотношение Б/Ж/У. У детей первого года жизни возможно употребление женского молока или молочных смесей при соответствующем расчете рациона и строгом контроле уровня фенилаланина в крови. Для восполнения недостающего белка используются аминокислотные смеси с низким содержанием фенилаланина и высоким содержанием тирозина, у детей старшего возраста компенсация происходит за счет растительных продуктов. Большой недостаток данного способа лечения — низкий комплаенс, особенно у детей подросткового возраста. Но при хорошей приверженности пациентов к диете снижение IQ можно свести к минимуму. Некоторыми исследователями были получены данные об эффективности применения гликомакропептидов в диете. Гликомакропептиды (GLP, glycomacropeptides) — белки, получаемые из молочной сыворотки, которая богата валином, изолейцином, треонином и при этом содержит низкий уровень фенилаланина. Их использование позволило бы сделать гипофенилаланиновую диету более физиологичной, но для широкого применения необходимы дальнейшие исследования и подтверждение безопасности применения GLP в течение длительного срока.
- Заместительная терапия BH4. Из-за участия BH4 в нескольких важных реакциях у больных BH4-зависимой формой ФКУ даже при хорошем соблюдении гипофенилаланиновой диеты остается симптоматика заболевания. В таком случае, как только на втором этапе лабораторной диагностики и/или на этапе медико-генетической диагностики подтверждается диагноз BH4-зависимой ФКУ, больным проводится тест на потенциальную чувствительность к сапроптерину дигидрохлориду — синтетическому аналогу BH4.
Иные методы лечения, имеющие потенциал:
- Большие нейтральные аминокислоты (The LNAAs, large neutral amino acids). Как было указано выше, фенилаланин способен конкурировать с другими аминокислотами (тирозин, триптофан) при взаимодействии с переносчиком LAT1. Некоторыми авторами было предположено, что в слизистой кишечника имеется подобный механизм, и при увеличении концентрации LNAAs всасывание фенилаланина будет уменьшаться.
- Генная терапия. Этот метод лечения мог бы стать идеальным решением, но в данный момент был тестирован лишь на мышах и требует дальнейшей серьезной разработки.
- Энзимотерапия фенилаланинамиаклиазой (PAL, phenylalanine ammonia-lyase). PAL — это фермент растений и дрожжевых грибков, осуществляющий катаболизм фенилаланина по альтернативному пути с образованием транс-циннамата и аммиака. За три последних десятилетия на мышах изучалось влияние PAL, внедренного в организм животного различными путями, начиная от оральных и инъекционных препаратов вплоть до помещения в кишечник генномодифицированных амеб, но, как и в случае с генной терапией, этот способ лечения требует дальнейшего изучения и разработки.
Источники:
- Blau N. et al. Phenylketonuria. // Lancet. Vol 376 October 23, 2010: pp 1417-1427.
- Blau N. Genetics of Phenylketonuria: Then and Now. // Human mutation, Vol 37, No. 6, 2016: pp 508-515.
- Hafid N.A., Christodoulou J. Phenylketonuria: a review of current and future treatments. // Translational Pediatrics 2015, 4(4): 304-317.
- Skirlou E., Lichter-Konecki U. Inborn Errors of Metabolism with Cognitive Impairment Metabolism Defects of Phenylalanine, Homocysteine and Methionine, Purine and Pyrimidine, and Creatine. // Pediatric Clinics of North America, Vol 65, 2018: pp 267-277.
- Руководство по педиатрии / [под ред. А.А. Баранова и др.] — Т: Врожденные и наследственные заболевания / [под ред. П.В.Новикова] — М.: “Династия”, 2007.
- Е.С. Северин и др.. Биологическая химия — М.: ООО «Медицинское информационное агентство», 2008.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017. https://www.pediatr-russia.ru/news/recomend
Источник: https://medach.pro/post/1493
Фенилкетонурия
Фенилкетонурия – тяжелое наследственное заболевание, природа которого заключена в нарушении обмена аминокислот. Опасна тем, что тотально поражает ЦНС в результате дефицита фермента – фенилаланина, который отвечает за формирование умственной деятельности, и как следствие происходит нарушение умственного развития человека.
Причины данного заболевания вызваны генетическими факторами, поэтому фенилкетонурия наследуется. Но в то же время, родители, могут быть вполне здоровыми и не иметь проблем с обменом веществ.
Фенилаланин обеспечивает построение белка и способствует образованию гормона щитовидной железы — тироксина. И если он правильно не перерабатывается, у человека развивается слабоумие.
Спасти ребенка, который родился с фенилкетонурией возможно только при максимально раннем установлении диагноза и начале лечения. Раннюю диагностику можно провести при помощи неонатального скрининга.
Признаки
Заболевание может проявляться не сразу. Фенилаланин, который поступает с молоком матери в организм больного ребенка, а затем и с другой пищей, достигает критической концентрации и воздействует на головной мозг ребенка, нарушая, таким образом, его функционирование. Как правило, при отсутствии своевременного лечения, первые симптомы станут заметны в первые месяцы жизни ребенка.
К признакам фенилкетонурии у детей можно отнести:
- гипотонус мышц, вялость;
- снижение координации движений;
- судороги конечностей и непроизвольные движения;
- обильное срыгивание, рвоту.
Дети больные фенилкетонурией имеют схожий фенотип. Такие малыши очень миловидны, имеют белокурые волосы, голубые глаза, нежнейшую, почти прозрачную кожу.
А когда болезнь начинает прогрессировать, они теряют эту ангельскую миловидность, так как в организме происходят тотальные нарушения.
Телосложение больных становится непропорциональным, на коже появляются экземами, дерматиты, а лицо начинает напоминать идиопатическую маску.
Высокий процент риска у детей, оба родителя которых, являются носителями данного гена. В такой ситуации, фенилкетонурия наследуется с вероятностью 25%. А в случае, когда больной ген передается плоду лишь от одного родителя, он в 50% сам становится его носителем, не имея внешних проявлений патологии.
Болезнь может быть вызвана таким факторами как близкородственные браки, мутация гена в локализации 12 хромосомы, наследование генов ФКУ.
Симптомы
Характерно, что данное заболевание никак не проявляет себя при внутриутробном развитии плода. Ребенок рождается в срок, имеет нормальный вес, а первые 2 – 2,5 месяца после рождения ничем не отличается от здоровых сверстников.
Первым и настораживающим симптомом фенилкетонурии является частая рвота.
Больной ребенок плохо набирает в весе, не может удержать головку, не проявляет интереса к окружающему миру, а также становится сонливым и раздражительным. Такие проявления родители могут заметить в возрасте от 2 до 6 месяцев.
Наиболее характерный симптом фенилкетонурии — повышенная потливость ребенка, которая имеет крайне специфический мышиный запах. Также специалисты отмечают явное уменьшение черепной коробки, а также судороги.
К дополнительным признакам у детей можно отнести позднее прорезывание зубов, задержку двигательной активности и беспричинную агрессию.
Если на этом этапе не начнется лечение, то симптомы фенилкетонурии начнут проявляться все стремительнее и будут выражаться в диспропорциях тела, постоянном треморе конечностей, специфических позах. После 6 месяцев, такие дети уже весьма отстают в психическом развитии от своих сверстников.
Кроме указанных симптомов болезни можно указать еще синюшность конечностей, потливость, дермографизм (изменение в окраске при местном воздействии на нее). Часто у больных отмечаются, помимо указанных выше симптомов, наличие дерматита, артериальной гипотонии, тремора конечностей, частых запоров, нарушения координации движений.
Диагностика
Поскольку фенилкетонурия наследуется довольно часто, несмотря на то, что оба родители здоровы, ранняя диагностика данного заболевания является крайне важной. Она позволит избежать его прогрессирования, тяжелого развития, а также необратимых последствий.
Поэтому в роддомах на 4-5 день жизни доношенного ребенка, у него берется кровь для анализа. А у недоношенных детей – на 7 день. Данная процедура имеет название скрининг-тест крови на фенилкетонурию.
Эта процедура несложная, и предусматривает взятие капиллярной крови по прошествии часа после кормления. Данной кровью пропитывают специальный бланк, и если, концентрация превышает 2,2% фенилаланина в крови ребенка, его направляют на осмотр в медико-генетический центр.
Лечение
Когда в крови больного снижается концентрация фенилаланина, постепенно в рацион вводят продукты животного происхождения. Также можно добавлять разнообразные фрукты и сезонные овощи, животные и растительные жиры, а также углеводы. При фенилкетонурии лечение предполагает постоянный контроль содержания фенилаланина в крови. Организму малыша необходимо быть снабженным этой аминокислотой в необходимом объеме для его нормального развития и роста.
При соблюдении рекомендаций врачей, как правило, ребенок сможет перейти к обычной системе питания.
Лечение заболевания медицинскими препаратами направлено на устранении симптомов. Например, при фенилкетонурии прописываются лекарства, ориентированные на устранение судорог, а также такие, которые стимулируют интеллектуальную деятельность. Необходимым является назначение курса массажа и лечебной физкультуры. Также могут предлагаться уроки на развитие логики.
Поскольку фенилкетонурия наследуется, то данное заболевание находится в компетенции врача генетика. Поэтому, его диагностирование осуществляется генетиком и педиатром на основании необходимых исследований в комплексе с общей симптоматикой. А в лечении основную роль играет соблюдение правил питания.
Согласно статистике, фенилкетонурией заболевает один ребенок из 10 тысяч новорожденных. С целью профилактики, будущим родителям стоит пройти медико-генетическое консультирование. Как правило, в группе риска пребывают те семьи, у которых были родственники, страдающие от данного заболевания.
Источник: https://dolgojit.net/fenilketonuriia.php
Фенилкетонурия: лечение, причины, симптомы, признаки
В результате нарушения обмена аминокислот может возникнуть фенилкетонурия. Заболевание довольно редкое. Оно встречается у одного человека из 8000. Болезнь возникает из-за дефицита фермента, при помощи которого осуществляется превращение фенилаланина в тирозин.
А теперь остановимся на этом подробнее.
Что такое «фенилкетонурия»?
Фенилкетонурия — врожденное генетическое заболевание, которое связано с нарушением обмена аминокислот. Вещество поступает в организм человека в составе белковой пищи. В результате происходит накапливание фенилаланина и его токсичных производных. В результате наблюдается поражение ЦНС.
Патология характеризуется умственной отсталостью, когнитивными и поведенческими нарушениями. У детей наблюдается явное отсутствие желания и мысли сопоставлять всё вокруг и анализировать. Возможно появление пассивного интереса, но не более. Присутствует проблема с восприятием и адаптацией к социуму.
Постепенно страдает и эмоциональная сфера. Наблюдается неустойчивость поведения.
В большинстве случаев развивается классическая форма заболевания. При ней наблюдается снижение активности печеночного фермента. Он носит название фениланин-4-гидроксилаз.
Существуют и другие разновидности патологии. В 1% случаев развивается атипичная разновидность заболевания. Она появляется в результате мутации другого типа генов. Он отвечает за процесс кодирования ферментов. Наследование заболевания осуществляется в соответствии с аутосомно-рецессивным схемой.
Из-за присутствия метаболического блока происходит активация побочных путей обмена фенилаланина. Это приводит к накоплению токсичных производных его действия. В частности, ими являются фениломолочная и фенилпировиноградная кислоты.
При нормальном состоянии организма вещество практически не образуется. Кислоты оказывают влияние на работу ЦНС. В результате наблюдается нарушение белкового обмена, а также обмена липопротеидов и липопротеидов. Одновременно присутствуют расстройства в транспорте аминокислот.
Нарушается обмен серотонина и катехоламинов. Актуальным становится перинатальные факторы.
Дополнительно начинают образовываться ортофенилацетат и фенилэтиламин. В норме они также не вырабатываются. Возможно в присутствии лишь незначительного количества осадков.
Избыток приводит к нарушению метаболизма липидов. Процесс осуществляется в головном мозге. Всё это становится причиной прогрессирующей патологии интеллекта. Человек может достичь даже состояние идиотии.
Окончательно процесс развития патологии пока не ясен.
Как выглядит фенилкетонурия с фото
Люди, страдающие патологией, имеют специфическую внешность. У них всегда очень бледная кожа. Она практически белая. Подобное явление возникает из-за того, что какой-либо пигмент в ней практически отсутствует. Волосы пациента имеют светлый оттенок. Глаза голубые.
Череп может незначительно уменьшаться в размерах. Возможно отклонение роста от нормы. Нередко он немного снижается. Однако имеются и примеры пациентов, которые обладали совершенно нормальным ростом.
Чтобы лучше разобраться, как выглядит ребёнок с фенилкетонурией, рекомендуется ознакомиться с фото.
Первые признаки фенилкетонурии
У новорожденных детей, у которых присутствует заболевание, какие-либо специфические клинические проявления не наблюдаются. Первые признаки патологии можно заметить в возрасте 2-6 месяцев. В этот период начинается манифестация заболевания. Как только в организм ребёнка поступает белок молока, начинают возникать первые признаки. Они неспецифические. У ребенка наблюдаются:
- мышечная дистония;
- вялость;
- гипервозбудимость;
- постоянное срыгивание;
- судорожный синдром;
- немотивированное беспокойство.
Одним из самых первых признаков заболевания является упорная рвота. Однако его нередко воспринимают, как клиническое проявление пилоростеноза. К 6-месячному возрасту у ребенка наблюдается явное отставание в психомоторном развитии. Пациент становится менее активным.
Он перестает узнавать даже самых близких людей. Ребёнок не предпринимает попыток сесть или встать на ножки. Состав мочи и пота становится аномальным. Появляется характерный мышиный запах или аромат плесени.
Нередко родители замечают у детей появление активного шелушения кожи, дерматита и экземы.
Если больной не проходит лечение, у него будет выявлена микроцефалия. Зубы начнут прорезаться только после полутора лет, четко наблюдается задержка в развитии. К трем или четырем годам может возникнуть глубокая умственная отсталость и полное отсутствие речи. Исключение составляют лишь некоторые непонятные звуки.
Дети с таким заболеванием отличаются диспластическим телосложением. У них нередко выявляют пороки сердца врождённого характера. Возможно возникновение вегетативной дисфункции и хронических запоров. Для подобного ребенка характерно:
- семенящая походка;
- тремор верхних конечностей;
- гиперкинез;
- поза портного.
Все вышеуказанные первичные признаки характерны для классической формы заболевания. При атипичных разновидностях патологии может присутствовать повышенная возбудимость, сухожильная гиперрефлексия и высокая степень умственной отсталости. Постепенно заболевание прогрессирует. К двум-трем годам ребёнок может скончаться.
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог.
Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен.
В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги.
После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится.
Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.
Причины и профилактика фенилкетонурии
Заболевание возникает из-за резкого снижения или отсутствия активности фермента печени. Именно с его помощью происходит превращение фенилаланина в тирозин.
Патология возникает в результате мутации генов, отвечающих за кодирование ферментов. Повреждения затрагивают ген, находящиеся на 12 хромосоме. Родственные браки повышают возникновение аномалии. Заболевания наследуются.
С ним одинаково часто сталкиваются мальчики и девочки.
При помощи профилактических мер предотвратить появление заболевания нельзя. Родители могут лишь оценить риск появления патологии у ребёнка в процессе планирования беременности.
Для этого потребуется пройти консультацию с генетиком, который направит будущих родителей на прохождение специфических исследований. Особое внимание необходимо уделить процессу, если имеет место быть кровнородственные браки, а также в роду присутствовали лица, болеющие этим заболеванием.
Если родители являются носителzvb дефектного гена, риск появления ребенка с фенилкетонурией составляет один к четырём.
Лечение фенилкетонурии
Диагностика заболевания входит в программу неонатального скрининга. Его должны пройти все без исключения новорождённые. Тест проводится на 3-5 день жизни ребёнка.
Если новорождённый появился недоношенным, процесс повторяют на 7 день жизни. Исследование осуществляется с помощью забора капиллярной крови. Если обнаруживается гиперфенилаланинемии, ребенка направляют к детскому генетику.
Чтобы подтвердить или опровергнуть диагноз, проводятся следующие исследования:
- выявляется активность печеночных ферментов;
- выполняется МРТ головного мозга;
- проводится биохимический анализ крови;
- Осуществляется ЭЭГ;
- выявляется концентрация тирозина и фенилаланина в крови ребенка.
Специфического лечения заболевания не существует. Основополагающим методом для борьбы с патологией выступает строгое соблюдение диеты. Она ограничивает поступление белка в организм больного.
Для кормления грудного ребенка применяются специальные смеси. Они же используются и для детей старшего возраста.
Основы меню становится низкобелковые продукты, которыми являются фрукты, овощи и аминокислотные смеси.
В более старшем возрасте суть диеты состоит в отказе от современных сладостей. Статистика показывает, что такие продукты содержат большое количество консервантов и эмульгаторов. Нередко в них добавляют фениланин или аспартам, оказывающие губительное влияние на больных патологией. Под запретом оказываются и:
- мясо любой природы;
- животный белок в продуктах;
- любые молочные продукты;
- растительные белковосодержащие продукты (частично);
- рыба.
Специалисты ведут большие споры, касающиеся возможности употребления материнского молока новорождёнными. Сегодня существуют две теории. В соответствии с одной из них, лучше перевести ребёнка на аминокислотные смеси, которые не содержат фенилаланин.
Приверженцы другой теории настаивают, что грудное молоко необходимо для нормального роста и развития ребёнка. Оно стимулирует иммунную систему, укрепляет связь между матерью и ребенком. Однако в этом случае женщина должна сильно скорректировать свой рацион. Избирательно нужно подходить даже к витаминным комплексам.
Нужно помнить, что риск обострений заболевания возрастает при малейших отступлениях от правил.
Обычно употребление вещества ребенком ограничивают до начала гормональной перестройки. В период полового созревания проблема может исчезнуть самостоятельно. Однако известны случаи, когда заместительная терапия и диета требуются пожизненно.
Если питание ребенка скорректировано до 8 недели жизни, это способно дать наибольший эффект. Если диетические ограничения были введены после 2 лет, это позволит лишь снизить выраженность симптомов.
Дополнительно может быть назначено употребление витаминов группы B, ноотропных средств, минеральных соединений и антиконвульсантов. В состав терапии в обязательном порядке должна входить лечебная физкультура, массаж и иглорефлексотерапия.
При атипичной форме заболевания диета пользы не приносит. Патология с ее помощью коррекции не поддается. В этом случае врачи назначают прием гепатопротекторов и противосудорожных средств. Такое лечение дает возможность облегчить состояние ребенка.
Дополнительно могут применяться ферментосодержащая терапия. В первую очередь врач назначает фенилаланинлиазу (PAL). Иногда принимается решение о необходимости использования кофаторама и его аналогов. Это фермент, позволяющий наладить процесс утилизации фенилаланила. База рассчитывается в соответствии с состоянием здоровья пациента, возможным прогнозом и возрастом.
Источник: https://zdorrov.com/drugie/fenilketonuriya.html
Фенилкетонурия. Что это за болезнь, причины, симптомы, лечение
Фенилкетонурия — это болезнь, которая поражает одного на 8 тысяч новорожденных детей. Это заболевание имеет генетический фон и заключается в неправильном обмене одной из аминокислот, а точнее, фенилаланина. Следствием этого метаболическогозаболевания может быть серьезное повреждение головного мозга ребенка.
Какие у этого заболевания симптомы? Можно ли вылечить эту болезнь? Прочитайте самую главную информацию о фенилкетонурии.
Симптомы и причины
Фенилкетонурия является наследственным заболеванием. Чтобы оно перешло ребенку, оба родителя должны быть носителем этого гена. Если у одного из родителей имеется это заболевание, тогда ребенок будет только носителем гена. Конечно, в этом случае, он в будущем может передать его своему потомству.
Какие симптомы у фенилкетонурии?Как уже было упомянуто, эта болезнь заключается в проблемах с обменом фенилаланина, в результате чего он накапливается в организме больного. Это приводит к повреждению мозга и тяжелой умственной отсталости.
Основные симптомы заболевания:
— судороги,- напряжение и тремор мышц,- неадекватное поведение,- рвота,- кожные высыпания,
— характерный запах.
Лечение и диагностика
Лечение фенилкетонурии заключается в определении дозы фенилаланина, которую пациент может принять.
Допустимая доза изменяется (в зависимости от возраста и состояния здоровья ребенка), поэтому важны регулярные обследования, определяющие его уровень в организме.
Сначала их следует делать каждую неделю, но с течением времени они могут быть реже. Старше 7 лет исследование делается один раз в месяц.
Болезнь важно быстро диагностировать, так как фенилкетонурия действует мгновенно, а ее последствия являются необратимыми. К счастью, в большинстве стран, новорожденные проходят обследование на третьи сутки жизни. Это позволяет обнаружить заболевание и принять соответствующие меры, целью которых является обеспечение ребенку возможность для нормального развития.
Питание при фенилкетонурии
Строгий рацион — основа питания при фенилкетонурии, который разрабатывается совместно с врачом-диетологом. Прежде всего, необходимо знать, в каких продуктах содержится меньшее количество фенилаланина.
Среди разрешенных продуктов:— мед, сахар, масла.
В небольших количествах можно употреблять:
— фрукты, овощи,- сливочное масло, маргарин,
— рис.
Из меню следует исключить:
— рыбу,- мясные продукты,- яйца, сыр,- молоко и молочные продукты,
— хлеб, а также некоторые мучные изделия.
Взамен этих продуктов применяются специальные препараты, которые поставляют организму необходимые витамины и минеральные соли. На рынке имеются также готовые продукты, созданные для людей, страдающих фенилкетонурией. Диета должна длиться как минимум до 14 лет, пока происходит динамическое развитие организма.
Фенилкетонурия как наследственное заболевание требует от пациента терпения и обязательного соблюдения рекомендаций врачей. Только таким образом можно избежать повреждения мозга из-за избытка вредных для организма веществ.
Стоит помнить, что генетическое заболевание, как фенилкетонурия, это не приговор – быстрая постановка диагноза позволит вашему ребенку вести нормальную жизнь, конечно при условии соблюдения диеты.
Источник: https://zen.yandex.ru/media/id/5a1fd5914bf161ced890a8d5/5cee2cd0c57ced00ae10b11f
1.4.3. Фенилкетонурия
(классификация
дана по Mc
Kusik
V.A.,
1988)
ФЕНИЛКЕТОНУРИЯ
1.
Классическая
фенилкетонурия (ФКУ) описана А.
Folling.,1934
г.
Заболевание
наследуется
аутосомно-рецессивно
и вызвано мутацией гена, локализующегося
в длинном плече 12 хромосомы.
В
основе
болезни
лежит дефицит фермента фенилаланин -4-
гидроксилазы, обеспечивающего превращение
фенилаланина в тирозин. В результате
метаболического блока происходит
значительное накопление в тканях и
жидкостях больного организма фенилаланина
и таких его производных, как
фенилпировиноградная, фенилмолочная,
фенилуксусная кислоты, фенилэтиламин,
фенилацетилглютамин,
и др.
В
патогенезе ФКУ
имеют значение следующие механизмы:
-
Прямое токсическое действие на ЦНС фенилаланина и его производных;
-
Нарушение в обмене белков, липо- и гликопротеидов;
-
Расстройства транспорта аминокислот;
-
Нарушение метаболизма гормонов;
-
Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
-
Нарушение функции печени : диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота
классической
ФКУ среди новорожденных по данным
массового скрининга в среднем колеблется
от 1 : 5000 до 1 : 10000 по разным регионам
России.
ФЕНИЛКЕТОНУРИЯ
2.
Впервые
атипичная ФКУ описана I.
Smith,
1974 г.
Заболевание связано с дефицитом
дигидроптеридинредуктазы.
Заболевание
наследуется аутосомно-рецессивно.
Генный дефект локализуется в коротком
плече 4 хромосомы, участке 4р 15.3.
В
результате недостаточности
дигидроптеридинредуктазы нарушается
восстановление активной формы
тетрагидробиоптерина, участвующего в
качестве кофактора в гидроксилировании
фенилаланина, тирозина, и триптофана.
Частота
заболевания
составляет 1 : 100 000 новорожденных.
Рано
начатое лечение способствует нормализации
фенилаланина в крови, однако не
предупреждает появление клинической
симптоматики, которая развивается в
начале второго полугодия жизни.
Фенилкетонурию 2 называют диеторезистентной
ФКУ.
ФЕНИЛКЕТОНУРИЯ
3.
Этот
вариант болезни описал S.
Kaufman
в
1978 г. Заболевание наследуется
аутосомно-рецессивно и связано с
недостаточностью 6-пирувоилтетрагидроптерин
синтетазы, участвующей в процессе
синтеза тетрагидробиоптерина.
Развивающиеся при этом расстройства
сходны с нарушениями, наблюдаемыми при
ФКУ 2.
-
Частота
болезни
составляет 1 : 30 000 новорожденных. -
Фенилкетонурия
3 также диеторезистентна -
ДРУГИЕ
ВАРИАНТЫ ФКУ.
Эти
формы ФКУ связаны с нарушением
альтернативных путей обмена фенилаланина.
Формируется метилминдальная
ацидурия и парагидроскифенилуксусная
ацидурия.
МАТЕРИНСКАЯ
ФЕНИЛКЕТОНУРИЯ.
Заболевание
развивается у потомков женщин, страдающих
ФКУ и не получающих диету в зрелом
возрасте. Патогенез
мало
изучен, предполагается, что он сходен
с патогенезом остальных форм ФКУ.
Тяжесть
поражения плода коррелирует с уровнем
фенилаланина в плазме матери. Так
как эмбрион особенно чувствителен
к тератогенным воздействиям , рекомендуется
начинать диету еще до наступления
беременности.
В суточном рационе
использовать менее 15-20 мг/кг
фенилаланина. При это важно избегать
дефицита незаменимых аминокислот.
КЛИНИКА
ФЕНИЛКЕТОНУРИИ.
При
рождении больные фенилкетонурией не
отличаются от других новорожденных.
Манифестация ФКУ происходит обычно в
возрасте 2-6 месяцев.
Первыми
проявлениями болезни
служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- срыгивание, рвота;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
- судорожный синдром;
- заплесневелый, мышиный, волчий запах мочи и пота.
-
При
отсутствии лечения формируется задержка
статико-моторного и психоречевого
развития, умственная отсталость
достигает, как правило, глубокой
степени. -
Характерны
такие фенотипические особенности, как
экзематозные и себорейные сыпи,
гипопигментация кожи, волос, радужной
оболочки глаз. -
ДИАГНОСТИКА
ФЕНИЛКЕТОНУРИИ. -
ФКУ
может быть диагностирована на основе
обнаружения следующих признаков:
- стойкой гиперфенилаланинемии (более 240 ммоль/л);
- вторичного дефицита тирозина;
- экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
В
настоящее время, согласно приказу
Минздрава России № 316 от 30.12.93 проведение
неонатального скрининга на ФКУ стало
обязательным. Скрининирующие тесты
должны быть простыми, недорогими и
информативными. Этим требованиям
отвечают методы, используемые для
ранней диагностики ФКУ:
- микробиологический тест Гатри;
- метод флюоресцирующих антител (лабораторный комплекс “Флюороскан”, позволяющий проводить 800 проб в час);
- метод тонкослойной хроматографии .
-
Оптимальные
сроки обследования новорожденных —
доношенных, зрелых — 5-6 день жизни;
недоношенных, незрелых, больных —
10-14 день жизни. - Трактовка результатов:
-
1 группа
— уровень фенилаланина не превышает 200
ммоль/л
(1-3 мг%)
— норма;
2 группа
— уровень фенилаланина составляет
200-500
ммоль/л
(3-10 мг%) — гиперфенилаланинемия. В
эту группу входят дети с транзиторной
гиперфенилаланинемией, вследствие
незрелости ферментных систем печени и
больные ФКУ.
За данной группой проводится
наблюдение по следующему плану: если в
течение 6 недель при еженедельном
исследовании уровень фенилаланина
остается менее 500 ммоль/л, контроль за
уровнем фенилаланина крови проводят
до 1 года первоначально каждые 3 месяца,
а затем каждые 6 мес. жизни.
При уровне
более 500 ммоль/л назначается диетотерапия.
3 группа
— уровень фенилаланина превышает 500
ммоль/л
(более 10 мг%), диагностируется ФКУ и с
момента постановки диагноза назначается
диетотерапия.
В
настоящее время разрабатываются и
внедряются молекулярно-генетические
методы диагностики генного дефекта при
ФКУ. Прямая диагностика мутантного гена
проводится с помощью синтетических
олигонуклеотидных зондов, этот метод
пригоден для дородовой диагностики ФКУ
и выявления гетерозиготного носительства.
Помимо
молекулярно-генетического анализа,
выявление гетерозигот может осуществляться
биохимическими тестами после нагрузки
фенилаланином в дозе 25 мг/кг.
Диеторезистентные
формы ФКУ диагностируют при помощи:
- исследования биоптеринов мочи ;
- перорального нагрузочного теста с тетрагидробоиптерином (через 4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит резкое снижение и нормализация уровня фенилаланина в крови с одновременным повышением уровня тирозина);
- исследования активности дигидроптеридинредуктазы и 6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов, эритроцитах, гепатоцитах.
ЛЕЧЕНИЕ
ФЕНИЛКЕТОНУРИИ.
-
Главным способом лечения является диетотерапия, ограничивающая поступление в организм фенилаланина; приступить к ней нужно немедленно после установления диагноза.
-
Из
рациона больных исключаются:
молоко,
молочные продукты, творог, мясо,
мясные продукты, колбасы, рыба, яйца,
хлебобулочные изделия, фасоль, горох,
орехи, шоколад. -
Белковым
эквивалентом этих продуктов питания
становятся гидролизаты белка, либо
аминокислотные смеси, лишенные
фенилаланина: “Лофенолак”, “Фенилфри”
(США), “Берлофен”, “Апонти”, “Гипофенат”
у детей до 4-5 лет и “Нофелан” — у детей
старше 5 лет. -
В
пищевой рацион входят овощи, фрукты,
мед, растительное масло, безбелковый
хлеб. -
Наиболее
рационально отменять
диетическое лечение
в
возрасте 7-8
лет.
-
Препараты с промедиаторным действием:
- Наком (комбинация карби ДОФА и лево ДОФА) — доза 100-375 мг/сутки в течение 3-4 недель, перерыв между курсами 1,5-2 месяца.
- Лево-дофа — доза 10-15 мг/кг в сутки;
- 5-окситриптофан — доза 10 мг/кг сутки.
-
Диеторезистентные формы. Лечение включает назначение тетрагидробиоптерина — доза 10-20 мг/кг в сутки.
ПРОФИЛАКТИКА
ФЕНИЛКЕТОНУРИИ.
- Выявление гетерозиготных носителей;
- Внедрение программ массового скрининга новорожденных для раннего выявления ФКУ и своевременного назначения диетотерапии;
- Пренатальная диагностика.
Источник: https://studfile.net/preview/2487369/page:12/