- В клиническом течении болезни выделяют две формы: классическую и атипичную.
- Дебют заболевания приходится на возраст 3-6 лет
- Первый симптом, на который родители и окружающие обращают внимание – это нарушение ходьбы. Нарушение ходьбы возникает на фоне изменения тонуса в мышцах, появления скованности (ригидности) мышц
В последующем присоединяются размашистые, беспорядочные, отрывистые движения, которые могут быть разными по амплитуде и интенсивности – хореические движения. Хореические движения могут затрагивать любые части тела. Они проявляются гримасничаньем, хаотичным высовыванием языка, облизыванием губ, размахиванием руками или ногами, танцующей походкой и др. Человек не может контролировать движения. Другой вид нарушения движений — атетоидные движения, которые затрагивают преимущественно кисти рук и стопы. Пальцы рук и ног совершают медленные, стереотипные «червеобразные» движения, неподвластные контролю больного.
У части больных движения замедляются, появляется дрожание в руках, ногах, языке.
Для классической формы характерно появление оромандибулярной дистонии, которая приводит к нарушению речи, нарушению глотания и постоянным прикусам языка.
Оромандибулярная дистония клинически проявляется насильственными избыточными движениями жевательных мышц, мышц рта.
Человек постоянно совершает жевательные движения или открывание рта, может быть, наоборот – сжимание челюстей и плотное сжатие губ.
Практически всегда происходит поражение сетчатки глаз, что проявляется выпадением полей зрения, появлением слепоты в ночное время.
Нарушение памяти, внимания может быть разной степени выраженности. Классическая форма болезни протекает волнообразно: периоды прогрессирования сменяются периодами ремиссии, но общее ухудшение в состоянии наступает быстро.
Дебют заболевания приходится на более поздний возраст (до 30 лет). Для атипичной формы характерно медленное прогрессирование состояния. К первым симптомам относят нарушение речи по типу эхолалии – повторение чужих слов, которое человек не контролирует.
Появляется нарушение произношения звуков и слов — дизартрия. Утрачивается чувство равновесия в положении стоя. На поздних сроках болезни присоединяются психические расстройства в виде агрессивности, депрессии. Практически у всех больных интеллект снижен.



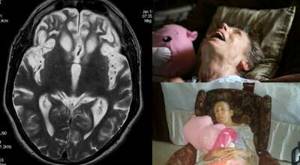
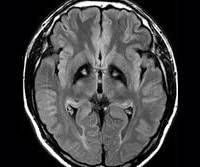
Особенности клинической картины
Признаки данной патологии могут варьироваться в каждом конкретном случае, и проявление симптомов зависит от формы болезни у индивида.
Детская форма Галлервордена Шпатца проявляется в возрасте между 5 и 10 годами. Считается классической формой болезни.
В подавляющем количестве случаев (90%) заболевание начинается с торсионной дистонии, которая поражает мышцы ног. У больного происходит сокращение мышц, затрудняется ходьба, изменяется походка. Затем поражаются мышцы лица, глотки и туловища.
Важно Пирацетам VS аналоги: что лучше выбрать?
Возможно присутствие блефароспазма, спазма кистей, спастической кривошеи, лицевого гемиспазма. Треть пациентов имеют мышечную ригидность, гипокинезию, тремор (признаки синдрома паркинсонизма).
К типичным признакам детской формы болезни также относят:
- эпилептический синдром;
- агрессивность;
- умственную отсталость (возникает из-за ухудшения памяти и внимания);
- асоциальность;
- зрительные нарушения (атрофия зрительных нервов, дегенерация сетчатки);
- психические расстройства.
Детская форма заболевания прогрессирует быстро, и полная потеря способности к передвижению наступает через 10-15 лет после дебюта болезни.
Подростковая форма развивается и протекает более медленно. В начале болезни проявляются:
- фокальная торсионная дистония (чаще всего затрагивает мышцы конечностей и челюстно-ротовые мышцы);
- нейропсихологические расстройства;
- поведенческие расстройства;
- интеллектуальные расстройства.
Атипичная форма болезни – наиболее редкая (15% случаев заболевания), и протекает отлично от первых двух форм. Для этой формы наиболее характерны такие симптомы:
- синдром паркинсонизма с невозможностью сохранять равновесие в положении стоя;
- непроизвольные движения в разных частях тела, дистония, хорея и атетоз (замедленные и порывистые движения), гемибаллизм (размашистые движения), миоклония (не длительные мышечные спазмы);
- деменция;
- эпилептический синдром;
- депрессивность, агрессия;
- патологические рефлексы, которые возникают вследствие разрушительных процессов в нейронах головного мозга.
Лечение
Лечение исключительно симптоматическое и назначается больному с учетом индивидуальных особенностей течения его болезни.
Специфического лечения не существует. Все лечебные мероприятия направлены на облегчение симптомов заболевания.
- Антиконвульсанты (Депакин, Конвулекс и др.) назначаются для снятия гиперкинезов – избыточных движений.
- Миорелаксанты (Баклосан, Мидокалм и др.) используются для уменьшения скованности мышц.
- Нейролептики (Клоназепам) и антидепрессанты (Циталопрам) — при психических нарушениях.
- Коррекция когнитивных нарушений (Глиатилин, Когитум и др.).
- Вазоактивные препараты (Винпоцетин и др.) улучшают обменные процессы в головном мозге.
- Ноотропы (Пантогам, Пирацетам и др.) стимулируют работу нервных клеток.
Симптоматическая терапия позволяет поддерживать относительное качество жизни человека в течение нескольких лет. Полная инвалидизация наступает через 10 и более лет.
Дифференциальная диагностика
БГШ по клинической картине напоминает многие другие заболевания нервной системы, основным проявлением которых является паркинсонизм.
- Болезнь Вильсона-Коновалова. Патология, связанная с нарушением обмена меди. Передается наследственно по такому же механизму, как и БГШ. Симптомы схожи: дистония мышц, деменция, дизартрия, дисфагия. Есть патогномичный симптом — необычный цвет глаз за счет колец Кайзера-Флейшера. Различают заболевания по результатам МРТ, анализа ДНК.
- Болезнь Гентингтона. Наследственное заболевание нервной системы, которое связано с атрофией стриатума (одна из структур базальных ядер) и коры головного мозга. Проявляется беспорядочными движениями и снижением когнитивных способностей. В отличие от БГШ, чаще появляется после 35 лет. Диагноз выставляется на основании генетического тестирования и МРТ.
- Болезнь Фара. Дегенеративное заболевание неясного происхождения. Как и БГШ, затрагивает базальные ганглии, только вместо железа в них происходит отложение кальция. На МРТ и КТ — симметричное поражение с кальцификатами. Клиническое проявление — паркинсонизм. Впервые симптомы могут появиться в любом возрасте.
Важно Кортикальная церебральная атрофия 1-5 степени: симптомы и лечение
Доврачебная помощь
Доврачебная помощь оказывается по следующему алгоритму.
- Поместить тепло (грелку, согревающий компресс) на поясничную область или применить горячую ванну, душ.
- Дать спазмолитики и анальгетики per os.
- Ввести достаточное количество жидкости (теплой воды, чая, минеральной воды) с учетом состояния сердечнососудистой системы.
- Ввести мочегонные препараты.
- Транспортировать больного в ЛПУ.
Примечание. Все мероприятия проводятся при отсутствии симптомов “острого живота”.
В.Дмитриева, А.Кошелев, А.Теплова
“Почечная или мочеточниковая колика, симптомы, первая помощь” и другие статьи из раздела Общая хирургия
- Мочекаменная болезнь
- Лечение почечной и мочеточниковой колики
- Вся информация по этому вопросу
Куда обращаться
Лечение
Каузальная (этиологическая) терапия неизвестна. Были попытки лечения энзимного дефекта. Хелаторы («ловушки») железа, такие как Дефероксамин, не оказывают эффекта, тем не менее, с 2007 года проводятся попытки проводить лечение хелатором железа Феррипрокс (Деферипрон).
В экспериментах на животных глубокая стимуляция мозга приводила к усилению дистоний и гиперкинезов. Гипокинезия может лечиться Леводопой, гиперкинезы — антихолинэргиками. Тем не менее, эффект Леводопы у пациентов с мутацией гена PANK2 очень сомнителен.
Для мышечной релаксации и купирования болевого синдрома часто назначается Баклофен или бензодиазепины.
Болезнь Галлервордена-Шпатца, PANK2 ч.м. – узнать цены на анализ и сдать в Москве
Интерпретация результатов
Метод определения Секвенирование. Выдаётся заключение врача-генетика!
- Исследуемый материал Цельная кровь (с ЭДТА)
- Доступен выезд на дом
- Исследование частых мутаций в гене PANK2.
- Тип наследования.
- Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания. PANK2 (PANTOTHENATE KINASE 2) – в большинстве случаев.
Ген расположен на хромосоме 20 в области 20p13 и кодирует пантотенат киназу 2 (Pantothenate kinase 2).
Определение заболевания.
Нейродегенеративное заболевание, связанное с накоплением железа в головном мозге с преимущественным накоплением в базальных ганглиях. Клиническая картина вариабельная.
Патогенез и клиническая картина.
Пантотенат киназа является необходимым регуляторным ферментом в биосинтезе коэнзима A. Она участвует в катализе цитозольного фосфорилирования пантотеновой кислоты (витамин В5), N-пантотеноилцистеина и пантотеина. Коэнзим A – это главный переносчик ацилрадикала, играющий центральную роль в метаболизме жирных кислот.
У пациентов с мутациями в гене PANK2 происходит накопление железа в клетках головного мозга, особенно в базальных ядрах. При патоморфологическом исследовании характерным признаком является гиперпигментация бледного шара и черной субстанции. Обнаруживается пигментация коры полушарий большого мозга и таламуса.
Важно Как и почему проявляется эпилептиформная активность на ЭЭГ
Пигмент находится внутри нейронов и глиальных клеток, расположенных около сосудов; содержит железо Классический вариант болезни Галлервордена-Шпатца манифестирует в детстве быстро прогрессирующей дистонией, паркинсонизмом, пирамидными знаками, дизартрией, ретинопатией и умственной отсталостью.
Перечень исследуемых мутаций может быть предоставлен по запросу.
- Болезни нервной системы. Руководство для врачей / Под ред. Н.Н. Яхно. — М.: Медицина, 2005. — Т. 2. — С. 170-171.
- Левин О.С., Юнищенко Н.А., Амосова Н.А. и др. Болезнь Галлервордена — Шпатца с поздним началом // Неврол. журн. — 2004. — № 2. — С. 36-46.
- Юдина Г.К., Шоломов И.И. Семейный случай болезни Галлервордена — Шпатца // Журн неврол. и психиатр. — 2003. — № 1. — С. 49-50.
- Hartig, M. B., Hortnagel, K., Garavaglia, B., Zorzi, G., Kmiec, T., Klopstock, T., Rostasy, K., Svetel, M., Kostic, V. S., Schuelke, M., Botz, E., Weindl, A., Novakovic, I., Nardocci, N., Prokisch, H., Meitinger, T. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann. Neurol. 59: 248-256, 2006.
- OMIM
- Специальной подготовки к исследованию не требуется. Обязательны к заполнению:
- *Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
- ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
- Специфическая картина при МРТ головного мозга.
- Кого надо обследовать при выявленной мутации:
- При выявлении у ребенка – обоих родителей, братьев и сестер.
- Интерпретация результатов
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
- Дифференциальная диагностика:
- болезнь Вильсона-Коновалова, паллидарная дегенерация.
- Результат исследования:
- Мутация не выявлена
- Мутация выявлена в гетерозиготном состоянии
- Мутация выявлена в гомозиготном состоянии
- Мутация выявлена в компаунд-гетерозиготном состоянии.
История редких нейродегенеративных заболеваний: Болезнь Пика, болезнь Галлервордена-Шпатца

Сегодня мы поговорим о двух редких нейродегенеративных заболеваниях – болезни Пика и болезни Галлервордена-Шпатца. Оба недуга названы в честь своих первооткрывателей, каждый из которых прожил сложную, но насыщенную жизнь.
Болезнь Пика – редкое хроническое прогрессирующее заболевание центральной нервной системы. Названа в честь Арнольда Пика (Arnold Pick), профессора психиатрии Карлова университета в Праге. Как и многие медицинские исследователи того времени, в самом начале своего пути он изучал широкий спектр дисциплин: клиническую неврологию, психиатрию и невропатологию, что в настоящее время довольно трудно представить. Современники говорили о нем, как об умном, скромном и принципиальном человеке, который внес существенный вклад в области психиатрии и неврологии: более 350 публикаций на немецком и английском языках тому подтверждение.
Во время своей работы в Карловом университете он столкнулся со множеством проблем, связанными с академическим преподаванием на немецком и чешском языках под контролем Австро-Венгерской империи. Зачастую ученому не хватало элементарных рабочих средств, он сталкивался с враждебностью и обидами, что мешало ему выполнять свой долг. Несмотря на это, его способностями к общению с пациентами с психозами и афазией восхищались, а вклад в медицину впоследствии оказался легендарным. Случаи больных с различными формами лобно-височной деменции и ее симптомами стали впервые описываться в конце XIX – начале XX века. В 1892 году Арнольд Пик изучал клинические симптомы пациента по имени Август Х., который страдал от тяжелой афазии – потери речи и слабоумия. Позже считалось, что именно Пик первым зарегистрировал случай первичной прогрессирующей афазии. В следующей работе немецкий доктор изучал случай пациентки по имени Анна Х., чьи симптомы включали в себя отклонения в поведении и потерю когнитивных функций, что также соотносилось с лобной деменцией. Третье исследование описывало проблему пациентки по имени Анна Д. – 75-летней женщины с ослаблением речевых навыков и потерей концептуальных знаний: Анна периодически забывала, как пользоваться привычными предметами домашнего обихода.
Все три случая легли в основу гипотезы Пика, что деменция является результатом локализованных ухудшений в конкретных областях мозга, а не общим нарушением функций головного мозга.
Впоследствии Алоис Альцгеймер, в честь которого немецкий психиатр Эмиль Крепелин (Emill Kraepelin) позже назовет одну из разновидностей сенильной деменции, выделил клубки белка, поселившиеся внутри нервных клеток в поврежденных участках мозга – тельца Пика.
Клетка Пика
Тельца Пика и клетки Пика содержат в себе аномальную форму тау-белка – органического вещества, присутствующего во всех нервных клетках, но по каким-либо причинам вырабатывающегося в ненормальных количествах или изменившего форму.
Во время многочисленных исследований ученым удалось установить причину аномального количества этого белка в нервных клетках: все дело в мутациях, затронувших хромосому под номером 17.
Но до сих пор науке неизвестно, почему меняется форма тау-белков.
Болезнь Пика можно было бы легко перепутать с болезнью Альцгеймера, если бы не тот факт, что первая затрагивает только определенные участки мозга, потому ее часто называют лобно-височной деменцией. Лобной доле головного мозга подконтрольны такие важные аспекты жизнедеятельности, как эмоции, речевое общение, поведение, торможение, а также некоторые формы движения. Очевидно, что любое изменение в клетках этой части мозга может привести к необратимым последствиям. Болезнь Пика – редкое заболевание, которое вызывает прогрессирующее и необратимое слабоумие. Человек с таким диагнозом испытывает трудности с контролем речи, поведением, мышлением, суждением и памятью. Как и у пациентов с другими типами деменции, могут возникнуть и резкие изменения личности. Чаще всего это расстройство диагностируют у людей в возрасте между 40 и 60, но иногда обнаруживается и у 20-летних. Универсального теста, определяющего лобно-височную деменцию, нет. Врачи могут провести ряд косвенных исследований: изучить историю болезни, провести тесты на устную и письменную речь, назначить детальное неврологическое обследование, а также рассмотреть ткани мозга при помощи МРТ и компьютерной томографии. Как лечится болезнь Пика? Не существует известных методов лечения, которые эффективно замедляют прогрессирование лобно-височной деменции, но некоторые препараты могут облегчить симптомы. Например, врач может выписать антидепрессанты или антипсихотические препараты для торможения эмоциональных и поведенческих изменений. Прогноз для людей с лобно-височной деменцией неутешителен. По данным Калифорнийского университета, расстройство прогрессирует в течение 8-10 лет. При этом от проявления первых симптомов до диагностирования заболевания может пройти несколько лет. В результате промежуток времени между установлением диагноза и смертью составляет около 5 лет.
Болезнь Пика встречается только в трех случаях на миллион, однако унесла достаточно жизней известных людей.
С этим расстройством в разное время боролись Роберт Флойд, известный своим вкладом в алгоритм Флойда-Воршалла, Дэвид Румельхарт – американский ученый, известный своими исследованиями научения и памяти в семантических нейронных сетях, Тед Дарлинг – выдающийся хоккеист, игравший за «Баффало Сейбрз», бенгальский поэт, писатель, музыкант и революционер Кази Назрул Ислам, бас-гитарист, композитор и художник, известный по своей работе с группой Van der Graaf Generator Ник Поттер и другие.
Дэвид Румельхарт Нейродегенерация с отложением железа в мозге первого типа или болезнь Галлервордена-Шпатца Юлиус Галлерворден (слева) и Уго Шпатц (справа)
 Юлиус Галлерворден (слева) и Уго Шпатц (справа)
Юлиус Галлерворден (слева) и Уго Шпатц (справа) Юлиус Галлерворден (Julius Hallervorden), чье имя связано с этим заболеванием, внес важный вклад в неврологию.
Несмотря на это, его деятельность во времена Второй мировой войны поднимает серьезные вопросы о моральных обязательствах медицинской науки.
Не существовало закона, обязывающего к участию в «Программе умерщвления Т-4» и проведению «усыпления», однако некоторые немецкие врачи шли на это практически добровольно. В их числе оказался и Юлиус Галлерворден.
Некоторые считают, что фамилию этого немецкого невропатолога нужно убрать из названия болезни.
Ученые предлагали переименовать недуг в «болезнь Марфы-Алмы», в честь двух сестер, чьи ткани головного мозга впервые исследовались Юлиусом Галлерворденом и Уго Шпатцем (Hugo Spatz).
В 1996 году Тодд Тейлор (Todd Taylor) предположил, что это расстройство можно описать как нейродегенеративное с накоплением железа в мозге (NBIA1), чтобы избежать нежелательного эпонима «Галлерворден-Шпатц». Сегодня во всем мире распространен именно вариант Тейлора.
Нейродегенерацию с отложением железа в мозге впервые описали двое немецких невропатологов – Юлиус Галлерворден и Уго Шпатц в 1922 году.
На открытие нового неродегенеративного заболевания их натолкнул случай одного семейства, в котором сразу у пяти сестер обнаружились признаки деменции и дизартрии – затруднения в произношении слов. После смерти сестер при вскрытиях врачи обнаружили коричневые пятна в различных областях головного мозга.
В особенно больших количествах их обнаружили в бледном шаре и черной субстанции – составных частях базальных ганглий. Этими загадочными пятнами оказались отложения железа.
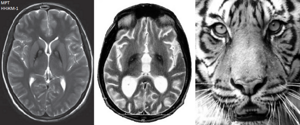
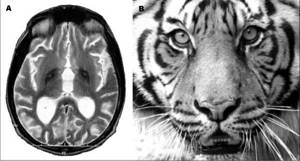
Дальнейшие исследования и описания произвел Мейер в 1958 году, который диагностировал 30 отдельных случаев NBIA1. Последователи Мейера описали пятерых пострадавших членов семьи и выдвинули гипотезу о том, что заболевание впервые возникло в центральной Европе. Таким образом они поддерживали вопрос о клинико-генетическом анализе. Болезнь Галлервордена-Шпатца является генетическим неврологическим расстройством, которое вызывает проблемы с движением. Обычно развивается в детском возрасте и с течением времени усиливается. У каждого больного симптомы могут различаться, однако выделяют общую группу признаков: искажения в сокращении мышц и трудности координации движения. Около четверти всех людей, страдающих от нейродегенерации этого типа, переживают нетипичную форму NBIA1, которая развивается после достижения 10-летнего возраста. В этом случае заболевание прогрессирует медленно и постепенно, в отличие от ситуации, когда недуг проявляется в более раннем возрасте. В то же время, взрослые больные в большей степени переживают значительное ухудшением речи, а также серьезные психические и поведенческие расстройства. Непреднамеренные, прерывистые мышечные сокращения на лице, теле или в конечностях также являются сигналом возникновения нейродегенеративного заболевания. Такие сокращения впоследствии могут изменять привычные для человека позы. К другим симптомам, сопровождающих больных, относят отвердение мышц, проблемы с координацией движения, припадки, дезориентацию, затруднения глотания, слабоумие и некоторые другие. Заболевание считается редким: на 1 миллион человек приходится до 3 больных. Чаще всего это члены одного семейства. После обнаружения первых симптомов врачи проверяют пациентов с помощью магнитно-резонансной томографии (МРТ). Главным маркером расстройства является наличие на снимке так называемого «глаза тигра» – расположенная в области бледного шара гиперинтенсивная зона овальной формы, окруженная еще большей гипоинтенсивной зоной. Последняя представляет собой, собственно, «глаз», а гиперинтенсивная – его «зрачок». Этот эффект, как правило, возникает из-за избыточного накопления железа.
Причина заболевания кроется в унаследованном дефекте в гене пантотенат-киназы 2 (PANK2). Белок PANK2 в организме человека контролирует выработку кофермента А, который, в свою очередь, помогает конвертировать жиры, некоторые аминокислоты и углеводы в энергию.
В медицинской практике встречаются случаи, когда нейродегенерация с отложением железа в мозге не вызвана мутацией гена PANK2. Ученые предполагают, что в таких случаях причиной болезни являются дефекты в одном или нескольких других генах, которые еще не идентифицированы.
В настоящее время NBIA1 не поддается лечению, и врачи лишь борются с ее симптомами. В зависимости от случая врачи назначают физиотерапию, медикаментозное лечение или оба решения одновременно. Физическая терапия уменьшает и предотвращает мышечную ригидность, спазмы и другие мышечные проблемы.
Одним из успешных методов борьбы с симптомами расстройства также является трудотерапия. С ее помощью пациенты развивают навыки для повседневной жизни и сохраняют текущие способности. Больные, обращающиеся к услугам логопеда, могут управлять расстройствами глотания или речевыми нарушениями.
Болезнь Галлервордена-Шпатца
О других заболеваниях на букву «Б»: Базилярная импрессия, Базилярная мигрень, Бери-бери, Беттолепсия, Боковой амиотрофический склероз, Болезнь Альцгеймера, Болезнь Вильсона, Болезнь Галлервордена-Шпатца, Болезнь Гамсторпа, Болезнь Гиппеля-Линдау, Болезнь Канавана, Болезнь Крейтцфельдта-Якоба, Болезнь Лафоры, Болезнь Мачадо-Джозефа, Болезнь мойя-мойя, Болезнь Мортона, Болезнь Паркинсона, Болезнь Пика, Болезнь Рефсума, Болезнь Фара.
Заболевание Галлервордена-Шпатца — прогрессирующее унаследованное расстройство, которое бывает ввиду отложений железа в головном мозге. Выражается болезнью Паркинсона, сбоем в умственном восприятии и в состоянии сознания, проявлением судорожного симптома, сокращением групп мышц, зрительными нарушениями.
Важным признаком наличия данной патологии является рисунок «глаза тигра» в виде округлой или линейной зоны в переднем мозге, который видно при проведении магнитно-резонансной томографии.
Терапия направлена на устранение симптоматики болезни средствами, применяемыми для лечения двигательных расстройств: препаратами для увеличения уровня домофина, медикаментами против эпилепсии.
Также лечение направлено на остановку судорог и подавление депрессивного состояния пациента.
В наше время заболевание неизлечимо и все действия направлены исключительно на ослабление негативных проявлений.
Данная патология встречается достаточно редко. После развития выраженных явлений патологии после бессимптомного течения отмечают три формы: детская, юношеская и взрослая.
До появления необходимой аппаратуры диагноз ставился исключительно специалистами по направлению «патологическая анатомия». После внедрения в медицинские организации необходимого оборудования появилась возможность ставить диагноз при жизни.
О причине накопления железа стало известно лишь в 2001 году. Выяснилось, что причиной всему служит повреждение гена, который и вызывает нарушение работы головного мозга.
После результатов данных исследований заболевание было переименовано в справочниках в пантотенаткиназа-ассоциированную нейродегенерацию.
Причины развития болезни
Заболевание Галлервордена-Шпатца появляется в связи с нарушением работы генов. Оно бывает и наследственным, и случайно приобретенным вследствие определенных мутаций. Генетической основой считают отклонение в гене пантотенаткиназы. Генетическим субстратом выступают нарушения в гене ферментов фотосинтеза, обозначенном на карте 20-й хромосомы 20 р 12.3 – р 13.
Выявлено порядка 50 мутаций. В конечном итоге такое генетическое повреждение приводит к уменьшению пантотенаткиназы, что ведет к сосредоточению серосодержащей кислоты. Данная аминокислота устанавливает прочное соединение с железом и пагубно воздействует на белки. В результате реакции происходит окисление, которое программирует гибель клетки.
На месте разрушенных клеток происходит размножение глиальных клеток. Такая необъяснимая реакция чаще всего затрагивает парную структуру переднего мозга и черное вещество, расположенное в области среднего мозга. Именно там и находят сконцентрированное железо, которое окрашено в коричневый цвет.
Так же в результате исследований были найдены образования, которые находились в мозговом веществе, коре мозга и в периферической нервной системе.
Симптомы болезни
Самая известная разновидность заболевания Галлервордена-Шпатца начинается в детстве в период с 4 до 10 лет. Самыми распространенными признаками болезни считаются:
- Проблемы с мышечным тонусом, особенно ног. Также случается, что больной занимает позы, которые выглядят неестественно. В таких случаях ребенку трудно ходить.
- Затем процесс затрагивает туловище, лицо и глотку.
- Так же с вышеуказанными проявлениями бывает дистония, которая наблюдается в нескольких частях тела. Бывает и появляется локальная дистония, непроизвольный спазм мышцы глаза, лицевой спазм, спастическая кривошея.
- Повышенный тонус мышц, при котором мышечная ткань становится плотной и возникает состояние малой двигательной активности с ограничением количества движений.
- Эписиндром.
- Психические нарушения, такие как снижение памяти и внимательности. Бывает в дальнейшем развивается умственная отсталость, именуемая олигофренией. Поведение таких пациентов может стать агрессивными и отклоняться от морально-нравственных норм.
- Нарушение произношения и зрения в результате атрофии нервов можно выделить как самостоятельный симптом у некоторых пациентов.
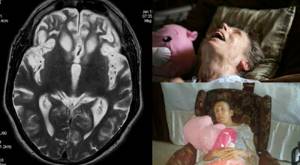 Проблемы с мышечным тонусом, особенно ног. Также случается, что больной занимает позы, которые выглядят неестественно. В таких случаях ребенку трудно ходить.
Проблемы с мышечным тонусом, особенно ног. Также случается, что больной занимает позы, которые выглядят неестественно. В таких случаях ребенку трудно ходить.Диагноз, поставленный в детском возрасте, обычно через 10-15 лет ведет к полной обездвиженности.
Юношеская форма патологии Галлервордена-Шпатца появляется в возрасте 10 лет и имеет замедленное течение. Также возникают проблемы с тонусом мышц, появляются непроизвольные сокращения, которые локализуются в конечностях, мышцах рта и челюсти. Присутствуют и типичные поведенческие особенности, связанные с интеллектом и психикой.
Взрослый вид недуга Галлервордена-Шпатца устанавливается уже после совершеннолетия. Выражается также неврологическими симптомами, которые характеризуются тремором и мышечным тонусом.
Выявляется недостаточная двигательная активность, болезненное состояние мышц, тремор и потеря координации движений.
Потерю координации легко можно выявить с помощью пробы Тавенарда — стоящего больного подталкивают вперед, тем самым выводя из равновесия. Врач обычно находится за спиной.
Главной особенностью течения заболевания считается совокупность неврологических признаков с другими отклонениями группы мышц. Категория нарушений умственных процессов может варьироваться от сохранности всех функций до прогрессирующего слабоумия.
Диагностика
Вспомогательными признаками данного заболевания могут служить патологические рефлексы, снижение умственных способностей, эписиндром, ухудшение зрения, атрофия сетчатки глаза, а также наличие в роду данного заболевания.
В целях диагностики проводят дополнительные исследования, в частности делают пациенту электроэнцефалограмму. Для постановки диагноза опираются на данные неврологического статуса и обследования, которое заключается в закономерности суммарной электрической активности мозга.
При явном ухудшении зрения требуется помощь офтальмолога для постановки диагноза, который проведет исследование по определению остроты зрения и осмотрит глазное дно специальными инструментами.
Генетик определяет наличие мутаций при составлении генеалогического древа, а также делает диагностику ДНК. Также с помощью дополнительного проведения ПЭТ выявляют сниженный обмен веществ в образовании серого вещества подкорковой части головного мозга.
Существует ряд других болезней со схожими патологиями, но в их клиническую картину не входит ряд особенностей заболевания Галлервордена-Шпатца. Схожие болезни по симптоматике: болезнь Хантингтона, Вильсона, Мачадо-Джозефа и другие болезни, связанные с генетически поражениями головного мозга.
Врачи при постановке диагноза основываются на данные, полученные в результате МРТ. На всех снимках пациентов наблюдается пигментированная область с железом. Изображение с однозначным видом данной болезни получило названия «Глаз тигра».
Более темные ткани этой зоны выглядят как глаз, а более светлые, яркие как зрачок. Момент появления «Глаза тигра» еще не определен учеными. По мнению одних он появляется еще до определенной симптоматики, по мнению остальных через некоторое время после яркого проявления болезни.
Лечебные прогнозы
К сожалению, помочь при данной болезни можно только блокируя симптомы, назначая препараты эффективные для лечения паркинсонизма. Попытки терапии, которые препятствуют накоплению железа хелатными соединениями («Дефероксамином») и антиоксидантами не получили успеха. В связи с этим применяется симптоматическая терапия с использованием трициклических аминоадамантанов. Но результаты проводимых исследований показывают отрицательное влияния препаратов на данную болезнь.
При конвульсиях применяют противоэпилептические препараты, такие как вальпроаты и бензодиазепины. Когда случаются болезненные судороги, то применяются миорелаксанты, такие как «Баклофен», «Толперизона гидрохлорид».
Когда случаются приступы эпилепсии, то топирамат или вальпроаты, при снижении памяти и умственных способностях применяют ипидакрин и холина альфосцерат. В случае серьезных психологических проблем назначают нейролептики, такие как «Рисперидон», «Кветиапин», «Клоназепам» и препараты против депрессии 3-го поколения: «Венлафаксин», «Циталопрам», «Дапоксетин».
Главной задачей врачей при выполнении симптоматической терапии заболевания Галлервордена-Шпатца является сокращение появления негативных симптомов, а также возможность позволить больным осуществлять уход за собой.
Наука не стоит на месте и ученые всего мира пытаются найти нужные лекарства для помощи пациентов. Идет активная оценка влияния на данное заболевание пантотеновой кислотой. Есть результаты исследований с эффективным влиянием метода, который позволяет неинвазивно стимулировать кору головного мозга.
Течение данной болезни тяжелое и прогноз зависит от стадии заболевания. Чем раньше эта болезнь началась, тем быстрее будут развиваться симптомы, приводящие к полной инвалидности через 10-15 лет. Самым положительным вариантом считается взрослая форма, чаще в тех случаях, когда слабоумие не ярко выражено. Длительность такого состояния может варьироваться до 20 лет.
Галлервордена-Шпатца болезнь
Болезнь Галлервордена-Шпатца (нейродегенеративное заболевание с аккумуляцией железа в мозге, neurodegeneration with brain iron accumulation-1, NBIA1)– редкое наследственное нейродегенеративное заболевание, при котором преимущественно поражаются базальные ганглии. Наследование аутосомно-рецессивное. Симптомы сильно варьируют от пациента к пациенту.
Классический вариант болезни Галлервордена-Шпатца манифестирует в детстве быстро прогрессирующей дистонией, паркинсонизмом, паримидными знаками, дизартрией, ретинопатией и умственной отсталостью.
У пациентов наблюдаются медленные спастические сокращения мышц губ, лицевых мышц и мышц туловища; хореоатетоз (непроизвольные, ненаправленные прерывистые мышечные сокращения), мышечная ригидность (необъяснимая тяжесть в мышцах), спастичность (внезапные, непроизвольные мышечные спазмы), атаксия (невозможность скоординировать движения), приступы затемнения сознания, дизориентации, ступор и деменция. Пациент становится привязанным к постели из-за гипокинезии и умирает в течении 10 лет после проявления первых клинических признаков заболевания. Тем не менее, приблизительно в 15% случаев, заболевание манифестирует во второй и третьей декадах жизни, а иногда и позднее и характеризуется медленным прогрессирующим течением и различными эктрапирамидными симптомами в совокупности с психиатрическими нарушениями. Более редкие симптомы — это болезненные мышечные спазмы, дисфазия, задержка умственного развития, гримасничанье, дизартрия.
При неврологическом обследовании обращают на себя вниманиеследующие симптомы: мышечная ригидность, слабость, насильственные движения, неестественная поза и тремор.
В наибольшем числе случаев, причиной заболевания являются мутации в гене, расположенном на хромосоме 20 (20p13-p12.3) и кодирующем пантотенат киназу 2 (Pantothenate kinase 2; PANK2 OMIM 606157). Пантотенат киназа является необходимым регуляторным ферментом в биосинтезе коэнзима A.
Она участвует в катализе цитозольного фосфорилирования пантотеновой кислоты (витамин В5), N-пантотеноилцистеина и пантотеина. Коэнзим A — это главный переносчик ацилрадикала, играющий центральную роль в метаболизме жирных кислот.
У пациентов с мутациями в этом гене происходит накопление железа в клетках головного мозга, особенно в базальных ядрах.
Специфического лечения для болезни Галлервордена-Шпатца нет. Лечение симптоматическое и поддерживающие. Показано положительное влияние витаминов: пантотеновой кислоты, коэнзим Q и другие антиоксиданты.
В Центре Молекулярной Генетики проводится прямая ДНК-диагностика болезни Галлервордена-Шпатца, основанная на поиске мутаций во всей кодирующей области гена PANK2.
Также возможно проведение секвенирования кодирующей последовательности только экзона 6 гена PANK2, в котором описаны 2 частые мутации G411R и T418M, на долю которых приходится 33% от общего числа поврежденных хромосом при данном заболевании.
Кроме того, для сходных клинических состояний проводится поиск мутаций в 15 генах, продукты которых ответственны за обмен меди и железа.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Болезнь Галлервордена-Шпатца
Галлерворден и Шпатц впервые описали заболевание в 1922 году, представив его как одну из форм наследственной дегенерации головного мозга, характеризующейся отложением железа в его тканях.
Термин «нейродегенеративное накопление железа 1 типа», или NBIA-1, стал использоваться чаще, чем термин «болезнь Галлервордена-Шпатца», однако в современной медицинской литературе и интернет-источниках распространены оба термина, и употребляют их одинаково часто.
Клиническая картина болезни Галлервордена-Шпатца
БГШ является генетическим (наследственным) заболеванием, для которого характерно прогрессивное неврологическое вырождение клеток мозга с параллельным скоплением железа в тканях. Ген заболевания находится на хромосоме 20 и участке хромосомы 20p13-p12.3.
Юлий Галлерворден и Уго Шпатц описали заболевание на примере пяти сестер, страдавших дизартрией (проблемами с речью) и прогрессирующей деменцией.
При вскрытии тел после смерти пациенток было обнаружено обесцвечивание отдельных участков головного мозга, в частности, бледного шара и черной субстанции.
Развивается болезнь по восходящему принципу — сначала жесткими становятся нижние, а затем и верхние конечности.
Отличительная особенность клинической картины – это непроизвольные движения, тремор (дрожание конечностей). Ослабевают мышцы горла и пищевода, отвечающие за глотание, артикуляцию, пережевывание пищи.
Со временем совершать жевательные и глотательные движения больным становится всё труднее. Также появляются серьезные сложности с координацией движений и регулированием положения тела в пространстве.
Кроме прочих нарушений возможно:
- ухудшение зрения;
- гипертонус мышц;
- истощение.
На поздней стадии заболевания появляются также серьезные отклонения психики. Как правило, болезнь Галлервордена-Шпатца развивается на первом или втором десятилетии жизни, то есть в возрасте 10-20 лет или старше.
После постановки диагноза пациенты живут обычно около 11 лет или чуть дольше. Смерть пациентов с данным расстройством наступает в возрасте 30 и более лет. Чем серьезнее степень развития заболевания, тем больше пациент нуждается в сторонней помощи. В последние годы жизни при утрате дееспособности рекомендована паллиативная помощь.
Причины болезни Галлервордена-Шпатца, факторы риска
БГШ является генетическим заболеванием, которое вызывается унаследованным дефектом в гене пантотенат киназы-2 (PANK2). Белок PANK2 контролирует образование в теле коэнзима А. Это соединение помогает организму конвертировать жиры, некоторые аминокислоты и углеводы в энергию.
В некоторых случаях болезнь Галлервордена-Шпатца может быть не связана с мутациями в гене PANK2. Медики полагают, что недуг вызывается дефектом в нескольких других генах. Однако идентифицировать эти гены специалистам пока не удалось.
Факторы риска
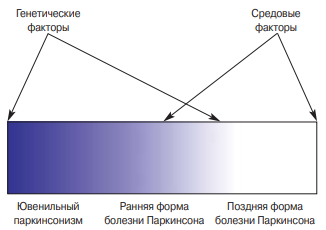
Основным фактором риска является наличие в семье родственников с данным расстройством. БГШ обычно проявляется впервые в детском возрасте, до 10 лет. Эта форма заболевания считается ранней. Поздняя форма характеризуется отсутствием симптомов в детском возрасте, но их проявлением после 20-летнего возраста.
Симптомы болезни Галлервордена-Шпатца, диагностика
Болезнь Галлервордена-Шпатца вызывает широкий спектр симптомов, которые варьируются в зависимости от тяжести заболевания и степени его прогрессирования.
Общий симптом — искажающие мимику и положение тела мышечные сокращения. Таким сокращениям могут быть подвержены:
- лицо;
- туловище;
- конечности.
Другие симптомы БГШ это:
- атаксия;
- судороги;
- скрюченные, спазмированные, жесткие конечности;
- отсутствие баланса;
- дезориентация в пространстве;
- бред;
- непоследовательность мыслей и поступков;
- ступор;
- слабоумие;
- замедленность жестов;
- недержание мочи;
- слюнотечение;
- затрудненное глотание;
- гримасы, нарушение нормального выражения лица;
- ригидность мышц;
- невозможность сформировать членораздельное предложение;
- болезненность при мышечных спазмах;
- неправильное положение тела.
Наиболее эффективными методами диагностики болезни Галлервордена-Шпатца являются:
- МРТ;
- физический осмотр;
- клиническая картина заболевания;
- генетическое тестирование (возможно не в любой клинике из-за сложности);
- семейная история болезни.
Лечение болезни Галлервордена-Шпатца
Лечение пациентов с болезнью Галлервордена-Шпатца остается в основном симптоматическим, то есть направленным на снятие симптомов, а не на устранение причины. Устранить генетическую аномалию, приводящую к БГШ, в настоящее время не представляется возможным.
Тремор эффективно устраняется дофаминергическими агентами. Антихолинергический препарат бензотропин может быть использован для смягчения ригидности мышц и уменьшения тремора, наравне с агонистами дофамина.
Для устранения дизартрии и слюнотечения рекомендованы:
- бромид метскополамина;
- логопедические манипуляции.
Системные хелатирующие агенты, такие как десферриоксамин, ранее использовались для удаления избытка железа из головного мозга, однако их эффективность не была доказана. Деменция является прогрессирующим симптомом, её прогрессирование можно замедлить, однако вылечить это состояние нельзя.
Рекомендуемые препараты при болезни Галлервордена-Шпатца:
- баклофен;
- бензтропин;
- мемантин;
- ривастигмин;
- донепезил.
Рекомендуемые занятия:
- физиотерапия;
- трудотерапия;
- логопедия
НЕЙРОДЕГЕНЕРАЦИЯ, ОБУСЛОВЛЕННАЯ НЕДОСТАТОЧНОСТЬЮ ПАНТЕТЕНАТ КИНАЗЫ
- НЕЙРОДЕГЕНЕРАЦИЯ, ОБУСЛОВЛЕННАЯ НЕДОСТАТОЧНОСТЬЮ ПАНТЕТЕНАТ КИНАЗЫ
- (ПКН, СИНДРОМ ГАЛЛЕРВОРДЕНА-ШПАТЦА)
- NEURODEGENERATION WITH BRAIN IRON ACCUMULATION 1; NBIA1; PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION; PKAN NEUROAXONAL YSTROPHY, JUVENILE-ONSET HALLERVORDEN-SPATZ DISEASE.
- MIM#234200
Генетика: мутации гена пантокинат киназы 2 (PANK2; MIM *606157). Ген пантотенат-киназы (PANK2) картирован на коротком плече хромосомы 20p13-p12.3 и экспрессируется преимущественно сетчатке и базальных ганлиях.
Тип наследования:
Эпидемиология:
Патогенез:Пантотенат киназа один их ключевых ферментов биосинтеза коэнзима А, катализирует в цитозоле клетки фосфорилирование пантотената (витамина B5), N-пантотеноилцистеина и пантотеина .
При мутациях в гене PANK2 происходит нарушение биосинтеза коэнзима А.
Коэнзим А играет важную роль метаболизме жирных кислот, однако, его роль в метаболизме и накопления железа в области базальных ганглиев остается до конца невыясненной.
Клинические проявления:Swaiman et al., в зависимости от возраста манифестации и характера течения ПКН выделяют две формы: классическую и атипичную. Для «классической» формы ПКН характерно раннее начало и быстрое прогрессирование неврологических нарушений. Клинические проявления заболевания у всех пациентов сходны.
Возраст начала болезни — от 3 до 6 лет. Первыми симптомами ПКН является нарушения ходьбы, возникающие в результате нарастающей мышечной дистонии и ригидности нижних конечностей. Нередким признаком заболевания является оромандибулярная дистония, что приводит к нарушениям речи, глотания и постоянной микротравматизации языка.
В 2/3 случаев отмечается пигментная дегенерация сетчатки, которая на начальных стадиях может проявляться в виде «ночной слепоты», в дальнейшем присоединяется выпадение полей зрения, и в некоторых случаях развивается амавроз. У 29% больных отмечаются когнитивные нарушения различной степени выраженности.
Акантоцитоз наблюдается в 3% случаев. Редко наблюдаются судороги. Для атипичной формы ПКН характерен более поздний дебют и медленное прогрессирование. Дебют заболевания приходится на первые три декады жизни (в среднем 13,6 лет). Начальными симптомами является нарушение речи в виде эхолалии , тахилалии или нарушение артикуляции.
На более поздних стадиях заболевания присоединяются психиатрические расстройства. У всех больных отмечается снижение интеллекта. Двигательные нарушения обычно отмечаются на поздних стадиях болезни. В некоторых случаях, заболевание напоминает болезнь Паркинсона.
В редких случаях у пациентов наблюдается пигментная дегенерация сетчатки и не встречается частичная атрофия зрительных нервов.
Диагностика:при МРТ головного мозга в Т2- взвешенном изображении обнаруживают симметричное снижение интенсивности сигнала в проекции бледного шара в сочетании с небольшими областями повышения сигнала внутри них («глаза тигра»). Основным методом подтверждения диагноза является ДНК анализ гена PANK2. Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru).
Лечение:Специфического лечения не разработано. Проводится симптоматическая терапия.
Прогноз
Скорость прогрессирования заболевания коррелирует с возрастом начала. Продолжительность жизни различная, большинство больных доживают до взрослого возраста.





